


Diterpene chemical space of Aeollanthus buchnerianus Briq. aerial part

Abstract
The Plectranthinae clade, which includes genera such as Plectranthus, Ocimum, and Aeollanthus, is well known for its diverse array of diterpenoids. While numerous studies have deepened the understanding of diterpene diversity across the clade, Aeollanthus species remain underexplored, with only two studies focusing on their diterpene profiles. The NMR-based chemical profiling of the EtOAc leaf extract of the rocky and succulent species Aeollanthus buchnerianus Briq. reveals a range of diterpenes with isopimarane and abietane skeletons including several previously unreported analogues. Interestingly, the isolated compounds provided insights into the breakdown patterns of both diterpene classes by examining the product ions in their MS2 spectra. These data offer valuable information for evaluating the taxonomic position of this species in relation to other species within the clade.Graphical Abstract
Keywords
Metabolomic Aeollanthus buchnerianus Isopimarane Abietane Structure elucidation Lamiaceae1 Introduction
Species of plants in the genus Plectranthus and in related genera in the family Lamiaceae including Coleus and Ocimum contain a range of well documented biologically active compounds [1, 2]. In contrast, other genera in the Plectranthinae clade like Aeollanthus [3] are not so well studied. For instance, a search using the keyword Aeollanthus in databases such as Scifindern and Reaxys® found only 50 and 52 publications, respectively. The genus contains about a hundred species distributed across sub-Sahara Africa excluding Madagascar and reputed for their resilience to harsh habitats like A. parvifolius [4] or for their strong fragrance and medicinal properties like A. suaveolens [5]. To date, most studies have focused on the composition of essential oils associated with insecticidal [6] and antimicrobial [7] properties.
Related genera often contain similar types of compounds often considered as signature molecules for the group. The Plectranthinae clade for instance are represented by diterpenoids as illustrated by the diversity of diterpenoids in Plectranthus [1]. With regard to Aeollanthus, only two studies have reported diterpenoids. In the late 90 s, Dellar et al. reported two abietane-type diterpenoids from A. buchnerianus [8]. More recently, Rijo et al. reported acyloxyisopimarane and other isopimarane diterpenoids from the aerial part of A. rydingianus [9]. One would deduce both isopimaranes and abietanes should represent markers of this genus too, but the hypothesis is yet to be tested.
Advances in mass spectrometry when combined with 1D NMR enable profiling of plant extracts for specific groups of compounds. A. buchnerianus in particular has not been investigated since the work of Dellar et al. [8]. It was therefore an opportunity to delve deeper into the profile of diterpenes in this species, resulting in the characterization of twenty diterpenes of which structures were confirmed following further NMR and MS analysis. Interestingly, the structures of eleven of the isolates were new to science including two isopimarane and nine abietane derivatives.
2 Results and discussion
The EtOAc leaf extract of A. buchnerianus was fractionated, for NMR-based chemical profiling, into 8 fractions (E1-E8) including a column wash. Each fraction was then submitted to NMR analysis, mainly 1H, 1H COSY, HSQC and HMBC, where each experiment was acquired beyond routine number of scans to allow minor components of the mixture to reveal their interactions. A relative consensus emerged after inspection of the major peaks informing on the class of compounds elicited by the plant and disclosing the main chemical characteristics of these compounds. Isopimaric acid (1) [10], 7-oxoisopimara-8, 15-dien-18-oic acid (2), 7-oxodehydroabietic acid (3) [11] and 14α-acetoxyabiet-7-en-18-oic acid (4) [8] were among the main structures one could extract from the difficult complexity of NMR data of the plant fractions. Thus, isopimaranes and abietanes were hypothesized as main diterpenoid classes that should be expected from the species, aligning with the report from Dellar et al. [8] where compound 4 was then isolated alongside a second abietane analogue not isolated herein. Interestingly, in both classes of diterpenoids, the identified major components of the fractions were shown to conserve the same oxidation to carboxylic acid of one of the geminal methyls at C-4. Accordingly, the fractions were thoroughly inspected looking for angular methyls in the HSQC and 1H, 1H COSY spectra that would correlate with a carbon at 175–185 ppm in the HMBC spectrum and would share with another methyl singlet, a HMBC interaction to a carbon at around 40–62 ppm for the position C-5 in both isopimaranes and abietanes. As a result, fractions E2, E3, E5 and E6 were selected for further analysis. The followed-up untargeted metabolomic analysis of those fractions by HPLC followed by NMR and LC–MS analysis led to the isolation and characterization of twenty diterpenes of which eleven were new chemistries (5–15) to science (Fig. 1).
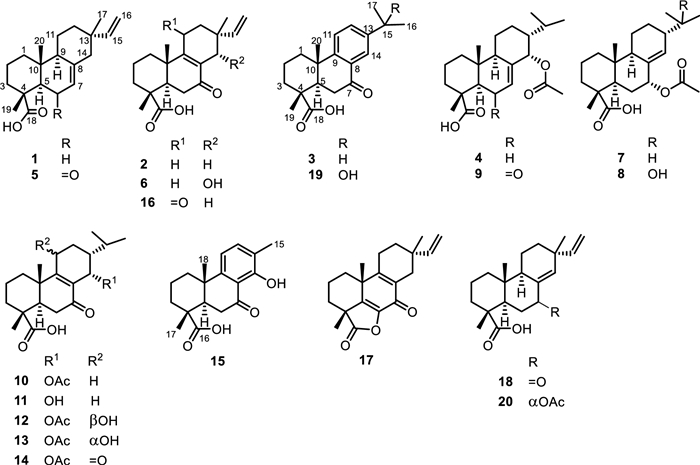
Structures of isolated compounds 1–20 from Aeollanthus buchnerianus
Compound 5 was colorless upon drying. Its negative-mode HRESIMS exhibited a deprotonated molecular ion peak [M-H]– at m/z 315.1961, corresponding to the molecular formula C20H27O3– (calcd for C20H27O3– m/z 315.1966). Its NMR spectra provided evidence of a vinyl group through the ABX system at δH 6.02 (dd, J = 10.6, 17.7 Hz, H-15)/ δC 149.1 (C-15), 5.16 (br s, H-16b) and 5.11 (br s, H-16a)/ δC 109.9 (C-16) and three angular methyls at δH 1.50 (s, H-19)/ δC 17.1 (C-19), 1.11 (s, H-20)/ δC 14.8 (C-20) and the allylic methyl at δH 1.05 (s, H-17)/ δC 21.7 (C-17), characteristics of (iso)pimaric acid derivatives [12] (Tables 1 and 2). As expected, both methyls H-19 and H-20 showed HMBC interactions (Fig. 2) to C-5 (δC 59.3) while the HMBC spectrum evidenced additional interactions from H-19 to C-3 (δC 37.5), C-4 (δC 42.4) and the carboxylic group C-18 (δC 179.3) and from H-20 to C-1 (δC 37.6), C-10 (δC 38.9) and C-9 (δC 51.6). Likewise, the HMBC spectrum exhibited cross peaks from the allylic methyl H-17 to C-15, C-13 (δC 36.3), C-14 (δC 45.3) and C-12 (δC 35.1). The relative downfield shift of C-5, as compared to C-5 in isopimaric acid [12], was indicative of a nearby ketone group positioned at C-6 (δC 198.1) and supported by HMBC cross peaks from an olefin at δH 5.86 (s, H-7) and H-5 to C-6. The relative stereochemistry of 5 was established based on NOESY interactions from H-19 to H-20 and from H-9 to H-5 (Fig. 2). Most importantly, the stereochemistry at C-13 was defined using the method developed in the literature and summed up by Seca et al. [12] in a review work where a clear distinction of pimaranes (αCH3−17) and isopimaranes (βCH3−17) were discussed based on 13C NMR resonances of the methyl C-17. Indeed, C-17 is expected to resonate at ~ 29 ppm in ∆8(14), 15 pimaradienes while it appears at ~ 26 ppm in ∆8(14), 15 and at ~ 22 ppm in ∆7, 15 isopimaradienes [12]. Therefore, compound 5 was an isopimarane as the methyl C-17 resonated at 21.7 ppm. Compound 5 was a new derivative with similar stereochemistry as compound 1 and characterized as rel−6-oxoisopimara-7, 15-dien-18-oic acid.
1H (400 MHz) NMR spectroscopic data for compounds of 5–15 (δ in ppm, J in Hz)
13C (100 MHz) NMR Spectroscopic Data for Compounds of 5–15 (δ in ppm)
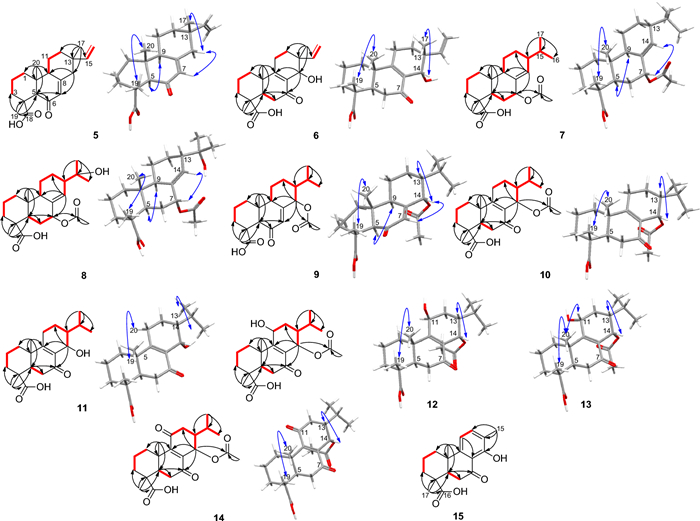
Selected COSY (red bold lines), HMBC (black arrows) and NOESY (blue double arrows) correlations in the new chemistries
Likewise, the negative-mode HRESIMS of compound 6 exhibited a deprotonated molecular ion peak [M − H]– at m/z 331.1909, supporting the molecular formula C20H27O4– (calcd for C20H27O4– m/z 331.1915). Its NMR spectra also evidenced resonances of a vinyl group through the ABX system at δH 5.64 (dd, J = 11.1, 17.7 Hz, H-15)/ δC 143.2 (C-15), 4.96 (m, H-16)/ δC 112.8 (C-16) and three angular methyls at δH 1.25 (s, H-19)/ δC 16.4 (C-19), 1.16 (s, H-20)/ δC 18.3 (C-20) and the allylic methyl at δH 1.08 (s, H-17)/ δC 24.0 (C-17), characteristics of (iso)pimaric acid derivatives [12] (Tables 1 and 2). Similar HMBC cross peaks, as in compound 5, supported a carboxylic group in compound 6 at C-18 (δC 179.4). The structure also exhibited a hydroxyl at C-14 through HMBC interactions (Fig. 2) from the allylic methyl H-17 to C-15, C-13 (δC 38.5), C-14 (δC 67.3) and C-12 (δC 27.9) and from H-14 to C-9, C-13 and C-7 (δC 199.8). Compound 6 was also classified as an isopimarane as the methyl C-17 resonated at 24.0 ppm. Further interactions were spotted in its NOESY spectrum from H-19 to H-20 and from H-14 to H-17 supporting the β-orientation of H-14 as depicted in the 3D representation of compound 6 (Fig. 2). Compound 6 was a new derivative with similar stereochemistry as compound 2 and characterized as rel−14α-hydroxy-7-oxoisopimara-8, 15-dien-18-oic acid.
Compound 7 was colorless upon drying. Its negative-mode HRESIMS exhibited a deprotonated molecular ion peak [M − H]– at m/z 361.2373, corresponding to the molecular formula C22H33O4– (calcd for C22H33O4– m/z 361.2384). Its 1H NMR and 1H, 1H COSY spectra exhibited a large spin system involving the methylenes H-11 (δH 1.80/1.28) and H-12 (δH 1.80/1.68), the olefin H-14 (δH 5.76), the methines H-15 (δH 1.57) and H-9 (δH 2.18), and the secondary methyls H-16 (δH 0.85) and H-17 (δH 0.88) (Table 1). Further resonances supporting the abietane core structure for compound 7 also included two additional angular methyls H-19 (δH 1.21) and H-20 (δH 0.80). As expected, both methyl singlets showed HMBC interactions to C-5 (δC 42.4) while the HMBC spectrum evidenced additional interactions from H-19 to C-3 (δC 36.9), C-4 (δC 46.7) and the carboxylic group C-18 (δC 184.5) and from H-20 to C-1 (δC 36.9), C-10 (δC 37.8) and C-9 (δC 48.2). The HMBC spectrum also exhibited cross peaks (Fig. 2) from the methine H-15 to C-16, C-17, C-14 (δC 134.3) and C-8 (δC 135.3). Interestingly, a second spin system, adjacent to the olefin as judged by HMBC interactions from H-7 to C-9 and C-14 and from H-5 and H-6 to C-8, was evidenced in the 1H, 1H COSY spectrum involving H-5, H-6 (δH 1.72/1.20) and H-7 (δH 5.29). Further cross peaks justified an acetyloxy group attached at C-7 from H-7 and an additional methyl singlet at δH 2.02 to the carbonyl at δC 170.5. The NOESY spectrum of 7 evidenced cross peaks from H-19 to H-20 and from H-5 to H-9. Most importantly, the spectrum exhibited interactions from H-7 to H-14 and from H-14 to H-13 informing on a planar orientation of H-7 and H-13 (Fig. 2). The axial positioning of the acetate at C-7 was further confirmed by the γ-shielding effect observed at C-5 as compared to compounds 1 and 4. Compound 7 was a new derivative with similar stereochemistry as compound 4 and characterized as rel−7α-acetyloxyabiet-8(14)-en-18-oic acid.
The negative-mode HRESIMS of compound 8 exhibited a deprotonated molecular ion peak [M − H]– at m/z 377.2325, corresponding to the molecular formula C22H33O5– (calcd for C22H33O5– m/z 377.2333), 16 Da away from compound 7. Indeed, like in 7, NMR resonances supported the abietane core structure for compound 8. However, compared to 7, the isopropyl at C-13 was replaced by a 2-hydroxypropyl as confirmed by HMBC cross peaks from the tertiary methyls H-16 (δH 1.16) and H-17 (δH 1.22) to an hydroxylated quaternary carbon at δC 72.5 (C-15) and the methine C-13 (δC 46.7). The relative configuration of compound 8 was also similar to that of compound 7 including the orientation of the acetate at C-7 also confirmed by its γ-gauche effect to C-5. Compound 8 was a new derivative with similar stereochemistry as compounds 4 and 7 and characterized as rel−7α-acetyloxy-15-hydroxyabiet-8(14)-en-18-oic acid.
Compound 9 was colorless upon drying. Its negative-mode HRESIMS exhibited a deprotonated molecular ion peak [M − H]– at m/z 375.2169, corresponding to the molecular formula C22H31O5– (calcd for C22H31O5– m/z 375.2177). Likewise, NMR resonances supported the abietane core structure for compound 9 including a carboxylic acid at C-18 (δC 179.2). The relative downfield shift of C-5, as compared to C-5 in 7-oxodehydroabietic acid (3), was indicative of a nearby ketone group positioned at C-6 (δC 198.8) and supported by HMBC cross peaks from an olefin at δC 6.08 (d, J = 2.8 Hz) and H-5 to C-6. Further cross peaks justified an acetyloxy group at C-14 supported by HMBC interactions from the methine H-15 to C-16, C-17 and C-14 (δC 73.3) and from H-14 and an additional methyl singlet at δH 2.06 to the carbonyl at δC 169.6. The relative stereochemistry of 9 was established as similar to that of compound 7 using the NOESY spectrum through same interactions from H-19 to H-20; from H-5 to H-9; from H-7 to H-14 and from H-14 to H-13 (Fig. 2). Compound 9 was a new derivative with similar stereochemistry as compound 4 and characterized as rel−14α-acetyloxy-6-oxoabiet-7-en-18-oic acid.
The negative-mode HRESIMS of compound 10 exhibited a deprotonated molecular ion peak [M − H]– at m/z 375.2168, corresponding to the molecular formula C22H31O5– (calcd for C22H31O5– m/z 375.2177). As in compounds 7–9, compound 10 was an abietane with an acetyloxy group at C-14 supported by HMBC interactions from the methine H-14 to C-9, C-8 (δC 130.7), C-7 (δC 196.5), C-15 (δC 27.9) and C-12 (δC 25.1); from the methine H-15 to C-16 (δC 20.2), C-17 (δC 21.7), C-14 (δC 63.6) and C-8 and from H-14 and an additional methyl singlet at δH 2.02 to the carbonyl at δC 169.6. The relative stereochemistry of 10 was established using the NOESY spectrum through interactions from H-19 to H-20 and from H-14 to H-13 supporting an α-orientation of the acetyloxy group (Fig. 2). Compound 10 was a new derivative characterized as rel−14α-acetyloxy-7-oxoabiet-8-en-18-oic acid.
The negative-mode HRESIMS of compound 11 exhibited a deprotonated molecular ion peak [M − H]– at m/z 333.2069, supporting the molecular formula C20H29O4– (calcd for C20H29O4– m/z 333.2071), 42 Da less than 10. Indeed, compared to 10, the HMBC spectrum of 11 lacks the cross peak from the oximethine H-14 to the carbonyl of ester characteristic of acetyl group. The relative stereochemistry of 11 was established as similar to that of compound 10 using the NOESY spectrum through interactions from H-19 to H-20 and from H-14 to H-13 (Fig. 2). Compound 11 was a new derivative with similar stereochemistry as compound 10 and characterized as rel−14α-hydroxy-7-oxoabiet-8-en-18-oic acid.
The negative-mode HRESIMS of compound 12 exhibited a deprotonated molecular ion peak [M − H]– at m/z 391.2116, corresponding to the molecular formula C22H31O6– (calcd for C22H31O6– m/z 391.2126). Compared to 10, the 1H NMR and 1H, 1H COSY spectra of compound 12 exhibited a large spin system (Fig. 2) involving two oximethines H-14 (δH 6.20) and H-11 (δH 4.78). Additionally, the HMBC spectrum exhibited cross peaks from both oximethines to C-9 and C-8 (δC 132.2); from H-14 to C-7 (δC 198.2), C-15 (δC 27.1) and C-12 (δC 30.7) and from the methine H-15 to C-16 (δC 20.0), C-17 (δC 21.7), C-14 (δC 63.6) and C-8. The absence of cross peak from H-11 to any carbonyl of esters justified positioning the acetyloxy group at C-14 of which characteristic signals were evidenced in the 1H and 13C NMR spectra at δH 1.98 (s, CH3CO)/ δC 21.2 and δC 169.6. The NOESY spectrum of 12 showed interactions (Fig. 2) from H-19 to H-20 and from H-14 to H-13 and H-17 supporting an α-orientation of the acetyloxy as in compound 10. However, the spectrum also exhibited important cross peaks from H-11 to H1−1 (δH 1.62)/H2−1 (δH 2.26) and H-12 not to H-20 nor to H-14, not enough to conclude on the orientation of HO-11.
Fortunately, eluted 8 min after 12 was compound 13 with the negative-mode HRESIMS exhibiting the same mass characteristics as 12. At first sight, its 1H NMR, 1H, 1H COSY and HSQC spectra align well with that of compound 12. Its HMBC and NOESY spectra in contrary disclosed some discrepancies much in the order-of-magnitude of some resonances. To start with, both secondary methyls H-16 (δH 1.03) and H-17 (δH 0.94) showed HMBC cross peaks (Fig. 2) to two methines at δC 30.6 for C-15 and 45.7 for C-13, downfield shifted by + 3.5 ppm and + 6.8 ppm, respectively, as compared to 12. Likewise, the angular methyl C-20 (δC 18.5) rather exhibited an upfield displacement of –1.7 ppm as compared to 12. The rationale behind all these shifts was suspected to relate to the orientation of the hydroxy group at C-11, mainly through δ- and γ-effects. Indeed, axial position of HO-11 eliminates the 1, 3-diaxial interaction of H-11 with H-13 leading to a gauche effect of the hydroxy group to C-13 (Fig. 3) as well as it also jams into the methyl C-20 causing its downfield shift. Both effects are less pronounced when HO-11 is equatorial as the chemical shifts of C-13, C-15 and C-20 become comparable to that of 10/11 that lacks the hydroxy group at C-11 (Fig. 3). Moreover, lateral hydroxy group at C-11 also favored its upfield shift of –1.5 ppm from the gauche interaction of the methyl C-20 (Fig. 3). Accordingly, the hydroxy group at C-11 was established as periplanar in compound 12 and oriented differently in compound 13. The orientation change of the hydroxy group in 13 was further supported by the clear interaction between H-11 (δH 4.54) and H-20 (δH 1.09), observed in the ROESY spectrum of 13. Compounds 12 and 13 were epimers at C-11 and new derivatives characterized as rel−14α-acetyloxy-11β-hydroxy-7-oxoabiet-8-en-18-oic acid and rel−14α-acetyloxy-11α-hydroxy-7-oxoabiet-8-en-18-oic acid, respectively.
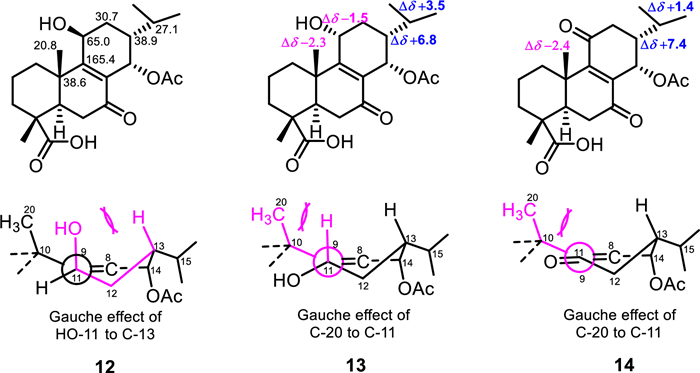
Chemical shift displacements and γ-gauche effects in compounds 12–14
The negative-mode HRESIMS of compound 14 exhibited a deprotonated molecular ion peak [M − H]– at m/z 389.1960, corresponding to the molecular formula C22H29O6– (calcd for C22H29O6–, m/z 389.1960). Compound 14 was the 11-oxo derivative of compounds 12–13 as its 13C NMR spectrum evidenced a new resonance of a ketone at δC 201.8 (C-11) and only one oximethine resonance at δC 63.6 (C-14). Additionally, the HMBC spectrum exhibited cross peaks from H-14 to C-9 (δC 156.4) and C-8 (δC 141.0), C-7 (δC 198.3), C-15 (δC 28.5) and C-12 (δC 40.1) and from the methine H-15 to C-16 (δC 19.8), C-17 (δC 21.1), C-14 (δC 63.6) and C-8. Further HMBC cross peaks justified an acetyloxy group attached at C-14 from H-14 and an additional methyl singlet at δH 2.07 to the carbonyl at δC 169.3. The NOESY spectrum of 14 showed interactions (Fig. 2) from H-19 to H-20 and from H-14 to H-13 and H-17 supporting an α-orientation of the acetyloxy as in previous compounds. Indeed, despite the carbonyl at C-11, compound 14 exhibits the same steric hindrances encountered in 13 in relation to the gauche effect of the methyl C-20 to C-11 and vice versa and justified by comparable chemical shifts of C-13, C-15 and C-20 in both compounds (Fig. 3). Compound 14 was a new derivative with similar stereochemistry as compounds 10 and 11 and characterized as rel−14α-acetyloxy-7, 11-dioxoabiet-8-en-18-oic acid.
Compound 15 was colorless upon drying. Its negative-mode HRESIMS exhibited a deprotonated molecular ion peak [M − H]– at m/z 301.1442, corresponding to the molecular formula C18H21O4– (calcd for C18H21O4– m/z 301.1445). Its 1H NMR spectrum exhibited two doublets of aromatic protons H-11 (δH 6.92) and H-12 (δH 7.48), and three methyl singlets H-15 (δH 2.29), H-17 (δH 1.42) and H-18 (δH 1.36) (Table 1). As expected, compound 15 also exhibits a carboxylic acid at C-16 as judged by HMBC cross peaks similar to those observed for compounds 5–14. Additionally, the HMBC spectrum evidenced cross peaks from the methyl H-15 to C-12, C-13 (δC 123.7) and C-14 (δC 160.5). A second spin system evidenced in the 1H, 1H COSY spectrum involving H-5 and H-6 (δH 2.97/2.46) justified a carbonyl at C-7 and supported by a chelated hydroxy proton at δH 13.16 (s, OH-14). The relative stereochemistry of 15 was established using the NOESY spectrum through interactions from H-19 to H-20. Compound 15 was a new derivative with similar stereochemistry as compound 3 and characterized as rel−14-hydroxy-7-oxo-16, 17-norabietic acid.
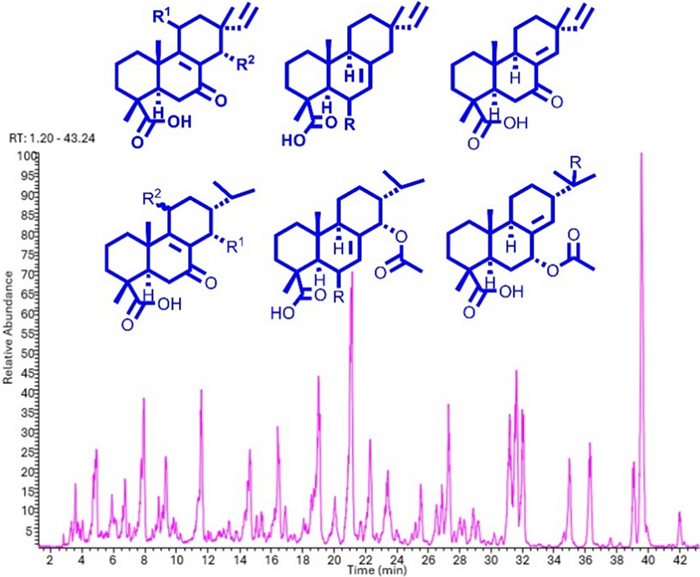
Further isopimaranes and abietanes were also isolated as known compounds including 7-oxoisopimara-8(14), 15-dien-18-oic acid (16) [10], dabeshanensin B (17) [13], 7-oxoisopimara-8, 15-dien-18-oic acid (18) [10], 15-hydroxydehydroabietic acid (19) [14] and 7α-acetoxysandaracopimaric acid (20) [15]. Wu et al. reported on the enantiomer of 2 [16]. The isolated compounds were used to annotate the LC-MSn base peak chromatogram of the studied EtOAc extract. Five of the major peaks in the chromatogram couldn't match any of the isolated characteristics (Fig. 4). These compounds could have been missed out during HPLC isolation as they could be not UV-active. However, the literature is not very eloquent on the fragmentation patterns of isopimarane or abietane derivatives. Thus, the isolated compounds 1–20 were used as standards in an attempt to define the breakdown logic of both classes of diterpenes that would allow the full annotation of the extract diterpenes.
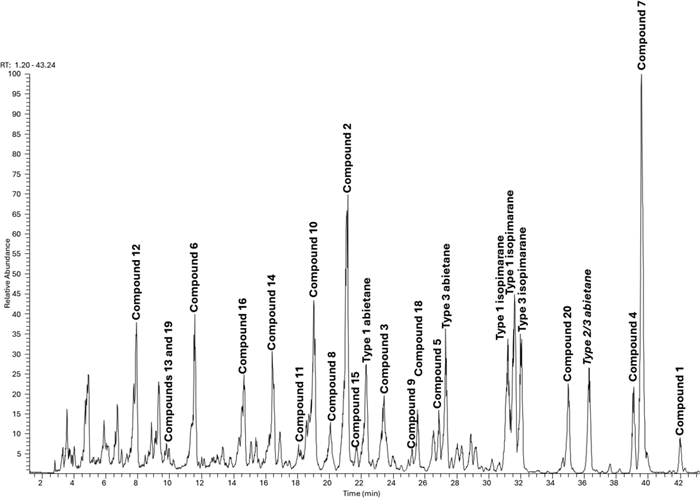
LC–MS base peak chromatogram of the EtOAc extract of A. buchnerianus
Isolated compounds were first clustered in three main types (Fig. 5) based on the olefin permutation within rings B and C, mainly ∆8(9), ∆7(8) or ∆8(14), irrespective of their diterpene classes. Some of the compounds could also be paired, compounds 6 and 11, as well as 7 and 20, as they share the same oxidation of the ring system but differ by the diterpene class they belong to. Other compounds from the same class were paired as they represent different skeleton types, such as compounds 9 and 10, 4 and 7, and 2, 5, and 18. Each of the pairs underwent LC-MSn analysis at a normalized collision energy of 40, and their product ions in MS2 spectra were compared to explore possible fragmentation patterns proper to diterpene types as well as classes by LC-MSn.
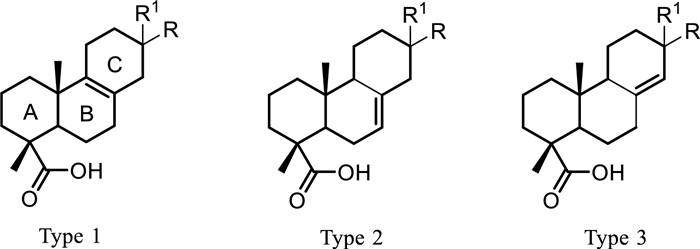
Diterpene types in isolated compounds
In this regard, the CID of the parent ion of compound 11 at m/z 333.2069 decays into a stable fragment ion at m/z 271.2067 (C19H27O–) following successive losses of H2O (m/z 315.1966) and CO2 (Scheme 1). Its isopimarane analogue, compound 6 (m/z 331.1909), also decays into a stable fragment ion at m/z 269.1910 (C19H25O–) after elimination of H2O and CO2 (Scheme 1). However, compound 6 alone unveiled a product ion at m/z 219.1391 (C14H19O2–) characteristic of a retro-Diels–Alder (RDA) fragmentation, leading to a neutral loss of m/z 68.0626 (C5H8) (Scheme 1), from the product ion at m/z 287.2016 (C19H27O2–) following CO2 loss from the parent [M − H]– ion. The collision of both compounds was then induced at an increasing energy level from 5 to 90. None of the product ions of compound 11 evidenced the RDA decay featuring the loss of m/z 68.0626. Instead, the MS2 spectra of compound 11 only evidenced an isopropyl loss as the energy increases from a CID of 50 (Scheme 1). Similarly, the decomposition of compound 2 begins with a decarboxylation, producing an ion at m/z 271.2070 (C19H27O–), which then decays following two pathways. The first one led to a RDA on ring C resulting in a neutral loss of C5H8, like with compound 6, while the other featured an α-elimination of methane, evidenced by the ion at m/z 255.1752 (C18H23O–). The decarboxylation of compounds 5 and 18 also produced a fragment ion at m/z 271.2070, which subsequently fragments further by releasing a neutral species of m/z 70.0786 (C5H10) instead.
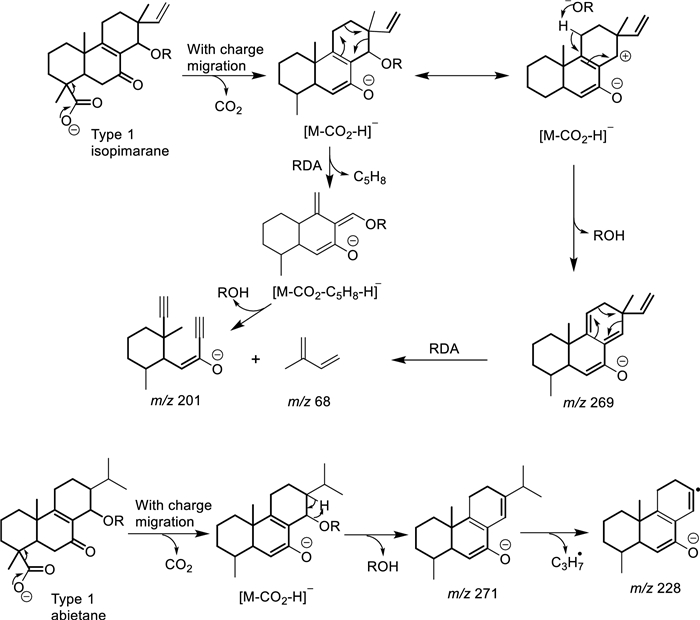
Retro Diels–Alder fragmentation mechanisms in Aeollanthus diterpenes
The retro-Diels–Alder fragmentation on ring C appears to separate isopimarane from abietane derivatives. This occurs in both classes of type 1 compounds when the C-14 position is oxidized to either an alcohol or an alcoholate group like in compounds 4, 6, 9 and 10–11. In such cases, the elimination of water or acetic acid extends existing conjugation in abietane analogues that prevents the RDA fragmentation from occurring (Scheme 1). The impossibility of the same elimination reaction in isopimarane derivatives results in a charge migration fragmentation, after the α-cleavage of hydroxy or acetate leading to the elimination of methylbutadiene by RDA (Scheme 1). In case position C-14 is not oxidized like in compounds 2 and 17, isopimarane derivatives of type 1 also undergo a neutral loss of methylbutadiene.
On the other hand, the CID of the parent ions of compounds 4 and 7 at m/z 361.2374 (C22H33O4–) showed similar product ions at m/z 301.2171 (C20H29O2–) after the α-elimination of acetic acid. However, in this case, the decarboxylation fragmentation reaction resulted in a neutral loss of formic acid, leading to the formation of an ion at m/z 255.2119 (C19H27–). The same decomposition was also observed in the case of compounds 1 and 20 of types 2 and 3. This represents a key feature in distinguishing between types 2 and 3 diterpenes and those of type 1. At an increase in energy level from 5 to 90, both compounds 4 and 7 could not be differentiated. Only their ability to undergo in-source release of acetic acid during ionization seems to differ. Compound 4 showed both the deprotonated ion at m/z 361.2374 and the in-source loss of acetic acid ion at m/z 301.2171 with comparable abundances while the latter was almost inexistant in the MS1 spectrum of compound 7 (Fig. S82). However, compound 9 with similar substitution as in compound 4 failed to show similar ability to in-source fragmentation.
The decarboxylation process and the releasing entity seem to differ with the diterpene types. In the case of types 1 and 3 with a carbonyl at C-7, CO2 is released through an α-cleavage with charge migration towards the carbonyl (Scheme 2) as portraited in the breakdown of compounds 2, 3, 6, 10–14, 15, 16 and 18. The decarboxylated entity and mechanism remain the same, except for the charge migration, when the carbonyl is located at C-6 like in type 2 compounds 5 and 9 (Scheme 2). However, in the absence of a carbonyl group at C-6 or C-7 like in compounds 1, 4, 7, 8 and 20, the decarboxylated body in diterpene types 2 and 3 is formic acid articulated here through a remote hydrogen fragmentation mechanism (Scheme 2).
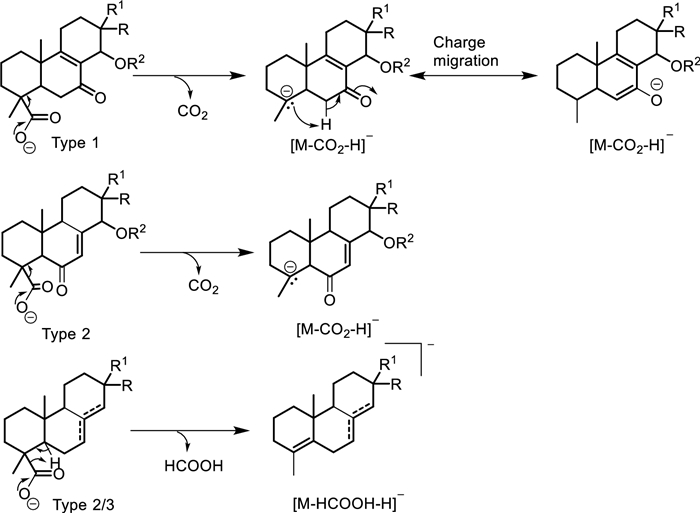
Decarboxylation reaction mechanisms in Aeollanthus diterpenes
Based on the two main features by which one should assign the olefin permutation type and the diterpene classes in A. buchnerianus, the five major peaks of the chromatogram missed during isolation were tentatively assigned diterpenes class and type (Fig. 4). Overall, irrespective of the diterpene types, the anticipated dissociation of CO2 from any of the CID of the parent ions of isolated compounds 1–20 was a second event, occurring after the breakdown of acetic acid, in acetylated derivatives and was prioritized otherwise. Moreover, none of the diterpenes were hydroxylated further or even glycosylated as none of the above patterns were identified in the 60% MeOH extract of the plant. The alignment of these characteristics with the chemistry as it develops in the plant could not be assessed comparatively given that only one chemical study of this plant exists to date.
Our findings also demonstrate that the diterpenes of A. buchnerianus possess distinct characteristics compared to those of Plectranthus and share greater similarity with analogues in Tetradenia, the two most extensively studied genera in the clade. Mainly, C-18/19 oxidized abietanes are rare in Plectranthus, and none of the abietane subclasses identified in Plectranthus were observed in A. buchnerianus [1, 2]. This contrasts with other genera within the clade such as Tetradenia which synthesizes royleanones, a major class of diterpenes commonly found in Plectranthus. The diterpenes of A. buchnerianus are more aligned with those described in Tetradenia. Both genera share similar diterpene classes, including abietanes and isopimaranes, as well as comparable double bond positions and oxidized carbons in the skeleton (C-6, C-7, C-14, and C-11) [17, 18]. The primary distinction lies in the extent of oxidation. In A. buchnerianus, the skeleton undergoes more advanced oxidation, particularly resulting in ketone groups at C-6, C-7, and C-11, while in Tetradenia, oxidation generally stops at the alcohol level [17, 18]. Worth mentioning as well, the diterpenes of this study are likely not enantiomeric knowing that enantiomeric diterpenes have not been reported from the Plectranthinae clade yet [1, 2, 17, 18]. The present results enrich the knowledge about the chemical diversity of diterpenoids in A. buchnerianus and open the opportunity to review whether other species in the genera contain similar compounds.
3 Experimental methods
3.1 General experimental procedure
LC–MS grade solvents (acetonitrile, methanol) and formic acid were obtained from Fisher Scientific (Loughborough, UK) and milliQ water was used for HPLC and LC–MS analysis. NMR spectra were acquired on a Bruker Avance-Ⅲ (1H NMR: 400 MHz and 13C NMR: 100.1 MHz) spectrometer equipped with a 5 mm cryoprobe. Chemical shifts were referenced to residual solvent signals and reported in parts per million (ppm). Spectra were processed using Bruker NMR academic Topspin software. Mass spectra were collected on a Orbitrap Exploris mass spectrometer, equipped with a Vanquish diode array detector (VH-D10) coupled to an Orbitrap Exploris 120 with a heated ESI source (Thermo Scientific, Germany), acquired in both negative and positive modes with a resolution of 60, 000 over m/z 125–1800 under various acquisition parameters like source voltages, sheath gas, auxiliary gas, sweep gas and capillary temperature set to 2.5 kV (negative mode) and 3.5 kV (positive mode), 50 (arbitrary units), 10 (arbitrary units), 1 (arbitrary units) and 350 ℃, respectively. Automatic MS–MS fragmentation was performed on top four ions of the TIC using an isolation width of m/z 2. High-energy C-trap dissociation with a normalized collision energy of 40 and an activation time of 0.1 ms was served to fragment ions. Collected data were inspected using Xcalibur v. 4.2.47 (Thermo Fisher Scientific). Chemical profiling of extracts was conducted on a Biotage® Isolera One system for splitting extracts into small fractions and a Waters Alliance 2695 HPLC system for isolation of compounds. A reversed-phase Disovery HS C-18 column (5 μm, 10 mm × 250 mm i.d., Supelco, UK) maintained at 35 ℃ served in compounds isolation and purification over gradient of acetonitrile + 0.1% formic acid (A) and water (B). The relative abundance of compounds in mixtures was evaluated via integration of isolated characteristic multiplets in the 1H NMR spectra.
3.2 Plant material
The whole plant of Aeollanthus buchnerianus was collected from the living collection at RBG Kew; accession No 1976–2374. The plant tissues were freeze-dried, milled to fine powders, and kept in the dark for further uses.
3.3 Extraction and isolation
Part of the milled plant materials (26.0 g) was serial extracted in solvents with increasing-polarities starting with n-hexane, then EtOAc and 60% MeOH affording dried extracts of 469.1 mg, 789.5 mg and 5.0 g, respectively. The EtOAc extract was split into seven small fractions (E1-E7) on the Biotage Isolera One system using a stepwise gradient of EtOAc in hexane starting from 100% hexane to 100% EtOAc, 10% increment and 3 column-volumes (CV) at each floor. The fractions were dissolved in CDCl3 and submitted to 1H NMR then to 2D NMR for chemical profiling. As rightly put above, only fractions E2, E3, E5 and E6 were further prepared for compound isolation. As E2 and E3 then E5 and E6 showed comparable compositions, they were combined to two main fractions E2 + 3 (110 mg) and E5 + 6 (106 mg). Fraction E2 + 3 was dissolved in 2 mL of ACN and injected into the Waters system, eluting with a constant flow rate of 2 mL/min of a linear gradient of acetonitrile (B) in water (A) (0–5 min, 35% B; 5–55 min, 35–55% B, 55–65 min, 100% B and 65–75 min, 35% B). Compounds were detected at 210, 254, 300 and 354 nm and collected by time into glass tubes. Cumulative fractions from eighteen injections of 100 μL each were collected and dried using a GeneVac concentrator (Genevac, Suffolk, UK). Likewise, fraction E5 + 6 was eluted with a linear gradient of acetonitrile (B) in water (A) (0–5 min, 35% B; 5–55 min, 35 to 55% B, 55–65 min, 100% B and 65–75 min, 35% B). Compounds were detected, collected and dried under the same conditions. Fraction E2 + 3 afforded 13 compounds including 12 diterpenes: 1 (4.5 mg), 2 (3.2 mg), 3 (1.6 mg), 5 + 17 + 18 (25:25:50, 1.1 mg), 4 (6.0 mg), 7 (11.5 mg), 9 (2.0 mg), 15 + 16 (36:64, 0.8 mg) and 20 (0.8 mg) whereas fraction E5 + 6 elicited 8 more diterpenes: 6 (1.9 mg), 8 + 11 (53:47, 0.8 mg), 10 (1.2 mg), 12 (2.4 mg), 13 (0.6 mg), 14 (2.6 mg), 19 (1.1 mg) and four miscellaneous. Compounds isolated as mixtures were not further purified.
3.4 rel-6-Oxoisopimara-7, 15-dien-18-oic acid (5)
Colorless oil; UV λmax 222, 259 nm; 1H and 13C NMR (CDCl3), see Tables 1 and 2; (−)-HRESIMS m/z 315.1961 [M − H]− (calcd for C20H27O3−, m/z 315.1966).
3.5 rel-14α-Hydroxy-7-oxoisopimara-8, 15-dien-18-oic acid (6)
Colorless oil; UV λmax 218, 245 nm; 1H and 13C NMR (CDCl3), see Tables 1 and 2; (−)-HRESIMS m/z 331.1909 [M − H]− (calcd for C20H27O4−, m/z 331.1915).
3.6 rel-7α-Acetyloxyabiet-8(14)-en-18-oic acid (7)
Colorless oil; UV λmax 200 nm; 1H and 13C NMR (CDCl3), see Tables 1 and 2; (−)-HRESIMS m/z 361.2373 [M − H]− (calcd for C22H33O4−, m/z 361.2384).
3.7 rel-7α-Acetyloxy-15-hydroxyabiet-8(14)-en-18-oic acid (8)
Colorless oil; UV λmax 200 nm; 1H and 13C NMR (CDCl3), see Tables 1 and 2; (−)-HRESIMS m/z 377.2325 [M − H]− (calcd for C22H33O5−, m/z 377.2333).
3.8 rel-14α-Acetyloxy-6-oxoabiet-7-en-18-oic acid (9)
Colorless oil; UV λmax 222 nm; 1H and 13C NMR (CDCl3), see Tables 1 and 2; (−)-HRESIMS m/z 375.2169 [M − H]− (calcd for C22H31O5−, m/z 375.2177).
3.9 rel-14α-Acetyloxy-7-oxoabiet-8-en-18-oic acid (10)
Colorless oil; UV λmax 218, 247 nm; 1H and 13C NMR (CDCl3), see Tables 1 and 2; (−)-HRESIMS m/z 375.2168 [M − H]− (calcd for C22H31O5−, m/z 375.2177).
3.10 rel-14α-Hydroxy-7-oxoabiet-8-en-18-oic acid (11)
Colorless oil; UV λmax 214 nm; 1H and 13C NMR (CDCl3), see Tables 1 and 2; (−)-HRESIMS m/z 333.2069 [M − H]− (calcd for C20H29O4−, m/z 333.2071).
3.11 rel-14α-Acetyloxy-11β-hydroxy-7-oxoabiet-8-en-18-oic acid (12)
Colorless oil; UV λmax 218 nm; 1H and 13C NMR (CDCl3), see Tables 1 and 2; (−)-HRESIMS m/z 391.2116 [M − H]− (calcd for C22H31O6−, m/z 391.2126).
3.12 rel-14α-Acetyloxy-11α-hydroxy-7-oxoabiet-8-en-18-oic acid (13)
Colorless oil; UV λmax 214 nm; 1H and 13C NMR (CDCl3), see Tables 1 and 2; (−)-HRESIMS m/z 391.2116 [M − H]− (calcd for C22H31O6−, m/z 391.2126).
3.13 rel-14α-Acetyloxy-7, 11-dioxoabiet-8-en-18-oic acid (14)
Colorless oil; UV λmax 214 nm; 1H and 13C NMR (CDCl3), see Tables 1 and 2; (−)-HRESIMS m/z 389.1960 [M − H]− (calcd for C22H29O6−, m/z 389.1970).
3.14 rel-14-Hydroxy-7-oxo-16, 17-norabietic acid (15)
Colorless oil; UV λmax 218, 266, 350 nm; 1H and 13C NMR (CDCl3), see Tables 1 and 2; (−)-HRESIMS m/z 301.1442 [M − H]− (calcd for C18H21O4−, m/z 301.1445).
Notes
Acknowledgements
The work was funded by a grant to MSJS from Procter and Gamble. The authors express their gratitude to the Kew Gardens horticulture team for caring for the Living Collection in the Gardens and providing access to the plant material.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Availability of data and materials
The NMR and LC–MS raw data generated during and/or analysed during the current study are available from the corresponding author on request.
Declarations
Competing interests
The authors declare no competing interests.
References
-
1.Grayer RJ, Paton AJ, Simmonds MSJ, Howes MJR. Differences in diterpenoid diversity reveal new evidence for separating the genus: Coleus from Plectranthus. Nat Prod Rep 2021;38: 1720-8. CrossRef PubMed Google Scholar
-
2.Gáborová M, Šmejkal K, Kubínová R. Abietane diterpenes of the genus Plectranthus sensu lato. Molecules 2022. CrossRef PubMed Google Scholar
-
3.Paton AJ, Springate D, Suddee S, Otieno D, Grayer RJ, Harley MM, Willis F, Simmonds MSJ, Powell MP, Savolainen V. Phylogeny and evolution of basils and allies (Ocimeae, Labiatae) based on three plastid DNA regions. Mol Phylogenet Evol 2004;31: 277-99. CrossRef PubMed Google Scholar
-
4.Ryding O. The genus Aeollanthus S. Lat. (Labiatae). Symb Bot Ups 1986;XXVI: 130-1. PubMed Google Scholar
-
5.Lupe FA, Lemes AC, Augusto F, Barata LES. Fragrant lactones in the steam distillation residue of Aeollanthus suaveolens Mart. Ex spreng and analysis by HS—SPME. J Essent Oil Res 2007;19: 271-2. CrossRef PubMed Google Scholar
-
6.Bohounton RB, Djogbénou LS, Djihinto OY, Dedome OSL, Sovegnon PM, Barea B, Adomou A, Villeneuve P, Tchobo FP. Chemical composition and the insecticidal activity of Aeollanthus pubescens leaf essential oil against Anopheles gambiae sensu stricto. Parasit Vectors 2021;14: 1-11. CrossRef PubMed Google Scholar
-
7.Martins RL, Simões RC, De Rabelo ÉM, Farias ALF, Rodrigues ABL, Da Ramos RS, Fernandes JB, Da Santos LS, De Almeida SSMDS. Chemical composition, an antioxidant, cytotoxic and microbiological activity of the essential oil from the leaves of Aeollanthus suaveolens Mart. Ex spreng. PLoS ONE 2016;11: 1-10. CrossRef PubMed Google Scholar
-
8.Dellar JE, Cole MD, Waterman PG. Unusual antimicrobial compounds from Aeollanthus buchnerianus. Experientia 1996;52: 175-9. CrossRef PubMed Google Scholar
-
9.Rijo P, Simões MF, Duarte A, Rodríguez B. Isopimarane diterpenoids from Aeollanthus rydingianus and their antimicrobial activity. Phytochemistry 2009;70: 1161-5. CrossRef PubMed Google Scholar
-
10.Chang LC, Song LL, Park EJ, Luyengi L, Lee KJ, Farnsworth NR, Pezzuto JM, Kinghorn AD. Bioactive constituents of Thuja occidentalis. J Nat Prod 2000;63: 1235-8. CrossRef PubMed Google Scholar
-
11.Tanaka R, Ohtsu H, Matsunaga S. Abietane diterpene acids and other constituents from the leaves of Larix kaempferi.. Phytochemistry 1997;46: 1051-7. CrossRef PubMed Google Scholar
-
12.Seca AML, Pinto DCGA, Silva AMS. Structural elucidation of pimarane and isopimarane diterpenoids: the 13C NMR contribution. Nat Prod Commun 2008;3: 399-412. PubMed Google Scholar
-
13.Hu CL, Xiong J, Gao LX, Li J, Zeng H, Zou Y, Hu JF. Diterpenoids from the shed trunk barks of the endangered plant: Pinus dabeshanensis and their PTP1B inhibitory effects. RSC Adv 2016;6: 60467-78. CrossRef PubMed Google Scholar
-
14.Yang XW, Feng L, Li SM, Liu XH, Li YL, Wu L, Shen YH, Tian JM, Zhang X, Liu XR, Wang N, Liu Y, Zhang WD. Isolation, structure, and bioactivities of abiesadines A-Y, 25 new diterpenes from Abies georgei orr. Bioorganic Med Chem 2010;18: 744-54. CrossRef PubMed Google Scholar
-
15.Esquivel B, del Socorro MN, Cárdenas J, Ramamoorthy T, Rodríguez-Hahn L. The pimarane-type diterpenoids of Salvia microphylla var. Neurepia Planta Med. 1989. CrossRef PubMed Google Scholar
-
16.Wu ZY, Zhang YB, Zhu KK, Luo C, Zhang JX, Cheng CR, Feng RH, Yang WZ, Zeng F, Wang Y, Xu PP, Guo JL, Liu X, Guan SH, Guo DA. Anti-inflammatory diterpenoids from the root bark of Acanthopanax gracilistylus. J Nat Prod 2014;77: 2342-51. CrossRef PubMed Google Scholar
-
17.Fernandez ACAM, Rosa MF, Fernandez CMMC, Bortolucci W, Melo UZ, Siqueira VLD, Cortez DAG, Gonçalves JE, Linde GA, Gazim ZC. Antimicrobial and antioxidant activities of the extract and fractions of Tetradenia riparia (Hochst.) Codd (Lamiaceae) Leaves from Brazil. Curr Microbiol 2017;74: 1453-60. CrossRef PubMed Google Scholar
-
18.Gazim ZC, Rodrigues F, Amorin ACL, De Rezende CM, Sokovic M, Teševic V, Vuckovic I, Krstic G, Cortez LER, Colauto NB, Linde GA, Cortez DAG. New natural diterpene-type abietane from Tetradenia riparia essential oil with cytotoxic and antioxidant activities. Molecules 2014;19: 514-24. CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2025.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
