


New semisynthetic α-glucosidase inhibitor from a doubly-chemically engineered extract

Ricardo L. E. Furlan and Mario O. Salazar would like to acknowledge for provided financial support by Universidad Nacional de Rosario (80020180300114UR and 80020180100128UR), CONICET (PIP Nº 11220200102423) and FONCYT (PICT2019-02232 and PICT2021-1034) for the development of this work
Abstract
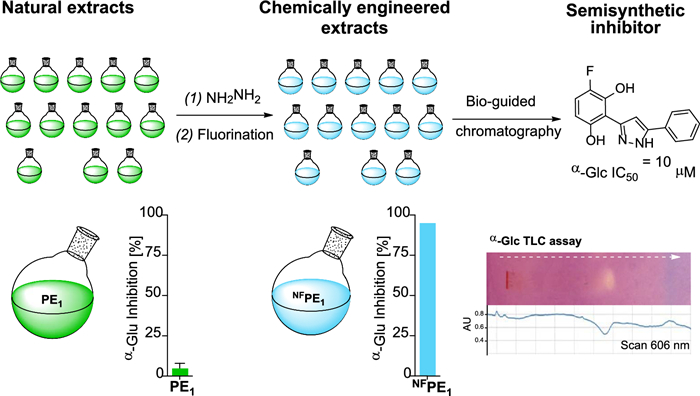
Chemically engineered extracts represent a promising source of new bioactive semi-synthetic molecules. Prepared through direct derivatization of natural extracts, they can include constituents enriched with elements and sub-structures that are less common in natural products compared to drugs. Fourteen such extracts were prepared through sequential reactions with hydrazine and a fluorinating reagent, and their α-glucosidase inhibition properties were compared. For the most bioactive mixture, a chemically modified propolis extract, enzyme inhibition increased 22 times due to the reaction sequence. Bio-guided fractionation led to the isolation of a new fluorinated pyrazole produced within the extract by chemical transformation of the flavonoid chrysin. The inhibitor results from the action of the two reagents used on four common functional groups present in natural products (carbonyl, phenol, aromatic carbon, and a double bond). The reactions led to the opening of a 6-member oxygenated heterocycle to produce a 5-member nitrogenated one, as well as the dehydroxylation and fluorination in two different positions of one of the aromatic rings of the natural starting material, all within a complex mixture of natural products. Overall, these transformations led to an approximately 20-fold increase in the α-glucosidase inhibition by the isolated inhibitor compared to its natural precursor.Graphical Abstract
Keywords
Natural products Chemically engineered extracts Glucosidase inhibitor Pyrazole Fluorine1 Introduction
The chemical engineering of natural product extracts can lead to semisynthetic compound mixtures with expanded diversity and improved biological properties [1-5]. Depending on the reaction protocol, the components of these chemically engineered extracts can be enriched in different halogens [6-9] or heteroatoms [10-18], and/or skeletons that are different from those of their natural precursors [1, 10, 11, 19-23]. Two reactions that have been successfully applied individually to modify natural mixtures are those with hydrazine [5] and with Selectfluor [7]. However, their sequential combination has not yet been explored.
Chemically engineered extracts (CEEs) have yielded various bioactive compounds, such as cytotoxic, [20, 23] antimicrobial, [6, 24, 25] and antiparasitic agents, [10, 26] as well as DNA-binding molecules [27] and enzyme inhibitors [7-9, 28], including some glucosidase inhibitors [11-13].
In humans, α-glucosidase (α-Glc) is a key digestive enzyme that catalyzes the hydrolytic cleavage of disaccharides (maltose and sucrose) into monosaccharides (glucose and fructose) in the intestine. Hence, the inhibition of α-Glc activity can retard the elevation of blood sugar, suppressing postprandial hyperglycemia, [29] and represents a useful strategy to control postprandial glucose level imbalance in type 2 diabetes patients [30]. Diabetes mellitus is among the top ten diseases in terms of impact on global health, [31] currently affecting approximately 9% of the adult population aged 20–79 years (425 million) [32]. Due to deficient insulin production (type 1 diabetes) or use (type 2 diabetes), patients experience abnormally high blood glucose levels which, if prolonged, can lead to severe cardiac, renal, cerebral, vascular, and visual complications [33-35].
Essential oils (EOs) are complex mixtures of natural hydrophobic small molecules derived from plant secondary metabolism [36, 37]. In nature, its production is diversity-oriented and regulates the interaction with other living organisms (plants, insects, or animals). EOs are increasingly recognized as valuable sources of molecules for drug discovery [37], exhibiting a wide range of biological activities [36]. There are only a few examples of chemically engineered essential oils, produced by halogenation [7, 8], sulfonylation [15], or thiocyanation [38], from which xanthine oxidase, tyrosinase, acetylcholinesterase, and glucosidase inhibitors have been identified.
Propolis are natural resinous mixtures produced by honeybees from substances collected from tree buds, exudates, and other botanical materials. Over the last two decades, they have gained significant scientific and commercial interest due to their broad clinical applications [39-41]. The chemical composition of propolis is highly complex, with over 500 compounds identified thus far, predominantly including terpenoids and phenolics (such as phenylpropanoids, flavonoids, lignans, coumarins, xanthones, etc.) [42, 43]. There are no reports on preparing chemically engineered propolis extracts (PEs).
The objective of this study was to explore whether the application of two sequential chemical reactions could lead to the identification of a new α-Glc inhibitor from the resulting chemically engineered extract. This investigation involved the chemical modification of 14 natural mixtures (essential oils and propolis extracts) through sequential reactions with hydrazine and Selectfluor® to introduce nitrogen and fluorine atoms into the natural molecular skeletons. While these reagents have been used individually in the past to create CEEs, their combined, sequential use is investigated here for the first time. Some of the resulting CEEs inhibited the enzyme α-Glc, and a fluorinated pyrazole was identified in the most active mixture. This compound, generated from the flavonoid chrysin within the extract, inhibits α-Glc more effectively than acarbose.
2 Results and discussion
2.1 Chemical diversification of natural mixtures and impact on α-Glc inhibitory properties
14 natural mixtures, 10 EOs and 4 PEs, were subjected to a reaction with hydrazine and, after removing the excess reagent, with Selectfluor®. The mass recovery for EOs ranged from 62 to 105% (average 94%) for the first reaction, 66% to 97% for the second reaction (average 80%), and 48 to 89% for the entire sequential process (average 70%) (Fig. 1a and Table S1). For PEs, the mass recovery ranged from 52 to 83% (average 67%) after the first reaction, 75% to 90% for the second reaction (average 84%), and 42% to 75% for the entire sequential process (average 57%) (Fig. 1a and Table S1).

a Mass recovery and b α-Glc inhibition at 3.125 µg/mL for NFEO1 (S. officinalis), NFEO2 (J. communis), NFEO3 (J. virginiana), NFEO4 (C. cassia), NFEO5 (T. occidentalis), NFEO6 (L. cubeba), NFEO7 (C. citratus), NFEO8 (A. absinthium), NFEO9 (E. caryophyllata), NFEO10 (C. odorata), NFPE1 (Esperanza, Santa Fe province), NFPE2 (Reconquista, Santa Fe province, PE2), NFPE3 (Lucas González, Entre Ríos province), and NFPE4 (Dean Funes, Córdoba province)
To evaluate their potential as a source of α-Glc inhibitors, the inhibition properties of the series of doubly chemically modified extracts were compared. When tested at 3.125 µg/mL, the CEEs from EOs exhibited enzyme inhibitions below 25%, whereas the CEEs from propolis showed inhibitions between 34 and 95% (Fig. 1b). The most interesting CEE of the series was the doubly modified extract from propolis sample number one (NFPE1) that displayed 95% inhibition (and close to 50% inhibition in a 1/16 dilution (0.781 µg/mL) (Fig. S1).
When compared with its precursor propolis extract (PE1), the chemically engineered NFPE1 produced significantly better inhibition at the three concentrations tested (12.5, 3.125, and 0.781 µg/mL) (Fig. 2a). This increased α-Glc inhibitory activity could be due to the presence of new semisynthetic compounds resulting from the incorporation of fluorine and/or nitrogen.
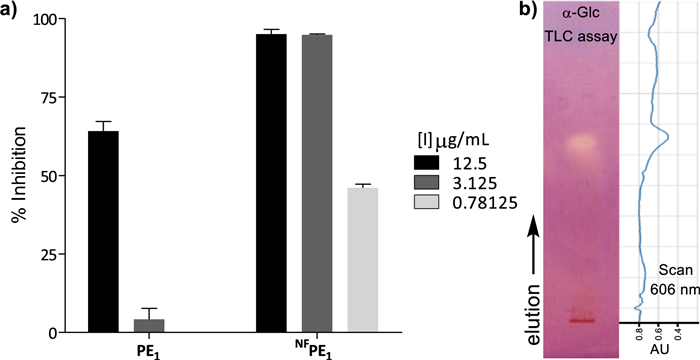
a α-Glc inhibition at 12.5, 3.125, and 0.781 µg/mL by PE1 vs. NFPE1 b TLC of NFPE1 revealed by α-Glc inhibition and scan of the TLC-plate at 606 nm (absorption wavelength of the diazonium dye). Silica gel, hexane–ethyl acetate, 1:1, V/V
It is worth mentioning that, since NFPE1 is a compound mixture of unknown composition, the observed α-Glc inhibition could result from the action of one good inhibitor, many weak inhibitors, or any other intermediate situation. These different scenarios define the real potential of a bioactive semisynthetic library as a source of hit compounds, so having this information beforehand facilitates early decision-making. Therefore, the α-Glc inhibition by NFPE1 was analyzed by effect-directed analysis on thin-layer chromatography (TLC), a technique particularly suited to assess the inhibition properties of compound mixtures [44-46].
This format assay allows measuring the inhibitory properties of a sample chromatographed on a thin layer, by covering it with a gel containing enzyme, substrate, and a revealing reagent for the product. For α-Glc, the TLC assay relies on the enzymatic hydrolysis of the substrate 2-naphthyl-α-D-glucopyranoside to form 1-naphthol, which reacts with Fast Blue B salt to produce a purple-colored diazonium dye [47]. Therefore, regions of the chromatogram containing α-Glc inhibitors appear as clear spots against the purple background and can be observed directly or by scanning the TLC at the dye absorption wavelength (606 nm). NFPE1 exhibited only one spot on the TLC-bioassay (Fig. 2b), suggesting that the inhibition observed for this mixture is due to one compound or a few compounds with similar chromatographic behavior.
2.2 Identification of an α-Glc inhibitor from the propolis doubly chemically modified extract NFPE1
Fractionation of NFPE1 using medium-pressure liquid chromatography (MPLC) bio-guided by the TLC α-Glc-inhibition assay led to the isolation of compound 3 (Scheme 1). This type of 3, 5-diaryl pyrazoles can be produced by the reaction of hydrazine with flavones through a nucleophilic attack of hydrazine at the C-2 followed by ring-opening and further nucleophilic attack of the second nitrogen atom at the carbonyl carbon and subsequent dehydration [4, 11]. In addition, hydrazine monohydrate can lead to the selective dehydroxylation of aryl pyrazoles with phloroglucinol-type substitution pattern on the A-ring [48]. According to this mechanism, the natural flavone chrysin (1), if present in the precursor propolis extract PE1, could have reacted with hydrazine to produce compound 2, which, upon fluorination, would lead to pyrazole 3 isolated from NFPE1 (Scheme 1).
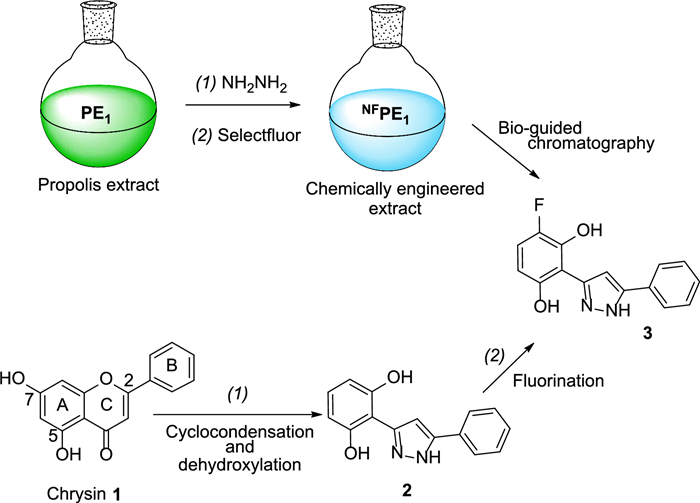
Reaction sequence that could lead to compound 3
2.3 Identification of the natural precursor of fluorinated pyrazol 3 and comparison of their inhibition properties
To corroborate the precursor-product relationship between chrysin (1) and pyrazole 3, firstly, the presence of the natural flavone was confirmed in the propolis extract used as the starting material (PE1) by HPLC, TLC, and 1H NMR (Fig. S2).
Secondly, pure chrysin was treated sequentially with hydrazine and Selectfluor® following the same procedure previously used for preparing NFPE1 from PE1 (Scheme 1). HPLC analysis of the reaction mixture showed the formation of the two expected pyrazoles 2 and 3 (Fig. 3), which were later isolated by MPLC in 28% and 15% yield, respectively. This confirmed that chrysin was the natural precursor of 3 (Scheme1).
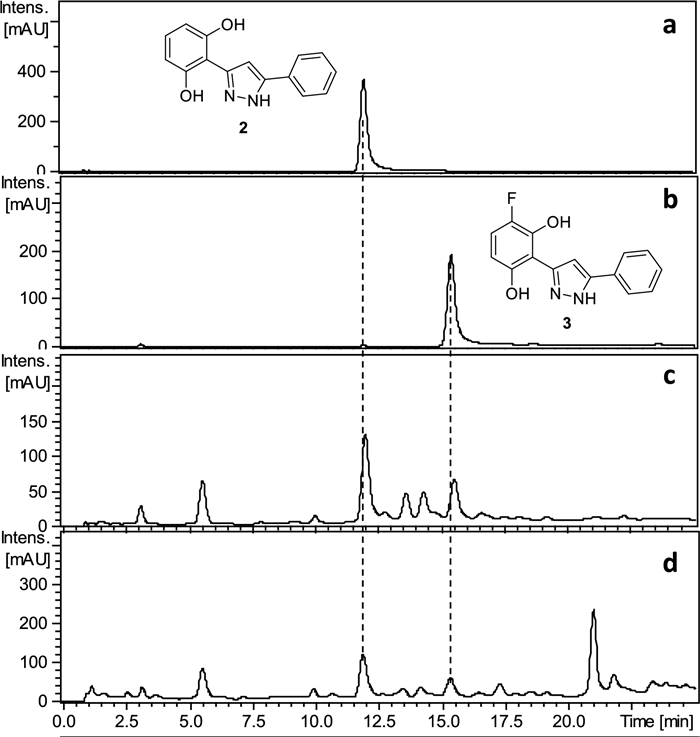
HPLC–UV-275 nm chromatograms of compound 2 (a), compound 3 (b), reaction mixture of the reaction with chrysin (c), and NFPE1 (d)
The α-Glc inhibition properties of chrysin and pyrazoles 1 and 2 were evaluated using a standard solution-based method. Chrysin inhibited α-Glc with an IC50 value of 212.30 ± 1.17 μM, which is more than 5 times higher than the value observed for the pyrazole 2 (IC50 = 38.27 ± 1.03 μM) and 20 times higher than the value observed for the fluorinated pyrazole 3 in the same experimental conditions (IC50 = 10.16 ± 1.23 μM). Furthermore, this fluorinated derivative (3) is twice more active than the reference α-Glc inhibitor acarbose (IC50 = 20.45 ± 1.02 μM). Physicochemical descriptors of compound 3 were computed to predict its drug-like properties using the SwissADME tool [49]. The compound demonstrated favorable drug-likeness, with no violations of the Lipinski [50], Ghose [51], Veber [52], Egan [53], or Muegge [54] filters. Additionally, no pan-assay interference structure (PAINS) alerts [55] or other structural alerts [56] were observed.
3 Conclusions
Two sequential reactions were applied to introduce nitrogen and/or fluorine into the natural components of a series of EOs and PEs. In some cases, the procedure led to a positive change in the inhibitory properties of the enzyme α-Glc by the extracts. One of the chemically engineered extracts (NFPE1) showed interesting α-Glc inhibition properties due to the presence of a fluorinated pyrazole. This compound resulted from the reaction of hydrazine with the B-ring of chrysin, which is present in the starting propolis extract (PE1), leading to the formation of the pyrazole ring. Additionally, hydrazine reacted with the A-ring resulting in dehydroxylation at position 7. Further, selective fluorination of the A-ring led to the formation of pyrazole 3, which inhibits α-Glc with a lower IC50 value than that of acarbose.
The results illustrate the potential of this strategy to generate and identify bioactive compounds. It is worth noting that in this particular case, the bioactive product is the result of the action of two reagents (hydrazine and Selectfluor®) on four common functional groups present in natural products (carbonyl, phenol, aromatic carbon, and a double bond), which led to the opening of a 6-member oxygenated heterocycle to produce a 5 member nitrogenated one, as well as the respective dehydroxylation and fluorination in two different positions of one of the aromatic rings of the natural starting material, all within a complex mixture of natural products. Overall, these transformations led to an approximately 20-fold increase in the α-Glc inhibition by both the whole compound mixture (4% inhibition by the starting natural PE1 versus 95% by the chemically engineered NFPE1 at the same concentration) and the isolated inhibitor (IC50 = 10 μM for the semisynthetic compound 3 versus IC50 = 212 μM for its natural precursor chrysin 1).
The process also highlights the utility of TLC-based inhibition assays to locate bioactive compounds within compound mixtures, by easily producing an α-Glc inhibition profile of the mixture before any purification step. This early information is useful as bioguide and facilitates the subsequent effect-directed purification of the corresponding inhibitor.
Overall, the approach suggests a shift in thinking: numerous semisynthetic compounds are generated simultaneously, and without knowing the exact outcome of the applied chemical process, bio-guided mixture selection and purification ensure that the most time-consuming work begins only after a promising compound has been detected.
4 Experimental
4.1 Preparation of CEEs
Since the exact composition of the natural mixtures used as starting material is unknown, some average properties of EOs or propolis components were used to estimate the appropriate amount of each reagent. The number of moles of reacting molecules was estimated considering 150 Da as the average molecular weight of EOs, 300 Da as the average molecular weight of propolis extract components, and one, as the average number of reacting group per molecule. Finally, 20 mol of hydrazine monohydrate and 1.1 mol of Selectfluor® per "estimated" mol of starting mixture components were used for the reactions.
Typical procedure for preparation of NFEOs: A solution of EO1 (100 mg, 0.66 mmol taking an average MW of 150 Da) and hydrazine monohydrate (645 μL, 13.32 mmol) in ethanol (5 mL) was stirred for 20 h under reflux. The reaction solution volume was reduced approximately to 1/3 under reduced pressure, water was added (5 mL), and the resulting solution was extracted with dichloromethane (DCM) (3 × 5 mL). The DCM fractions were dried (anh. Na2SO4), filtered, and evaporated at reduced pressure. The obtained crude (71.9 mg, 0.48 mmol) was dissolved in ethanol (5 mL), Selectfluor® was added (186.8 mg, 0.53 mmol), and the solution was stirred at room temperature for 20 h. Water was added (5 mL) and the aqueous solution was extracted with DCM (2 × 5 mL). The DCM fractions were dried (anh. Na2SO4), filtered, and evaporated at reduced pressure.
Typical procedure for preparation of NFPEs: A solution of PE1 (100 mg, 0.33 mmol taking an average MW of 300 Da) and hydrazine monohydrate (322 μL, 6.66 mmol) in ethanol (5 mL) was stirred for 20 h under reflux. The reaction solution volume was reduced approximately to 1/3 under reduced pressure, water was added (5 mL), and the resulting solution was extracted with DCM (3 × 5 mL). The DCM fractions were dried (anh. Na2SO4), filtered, and evaporated at reduced pressure. The obtained crude (51.5 mg, 0.17 mmol) was dissolved in ethanol (5 mL), Selectfluor® was added (66.90 mg, 0.19 mmol) and the solution was stirred at room temperature for 20 h. Water was added (5 mL) and the aqueous solution was extracted with DCM (2 × 5 mL). The DCM fractions were dried (anh. Na2SO4), filtered, and evaporated at reduced pressure.
4.2 Fractionation of NFPE1
The NFPE1 was chromatographed in MPLC-UV (Elldex-Alltech). 50.0 mg were directly loaded on a Latek model M2 (2 cm Id × 33 cm length) glass column filled with Silica gel 60 RP-18 (15–25 μm, LiChroprep RP-18, Merck). Mobile phase: 1% Formic acid solution and methanol. Method: 0–60 min 60% methanol, 100 min 100% methanol. Injection solvent: methanol. Flow: 4 mL/min. The effluent of the column was monitored at 254 nm. Fractions were automatically collected every two minutes to obtain fifty fractions (F1 − F50). The fractions F13-15 contained 1.5 mg of pure of pure fluorinated pyrazole 3 (3.0% final yield).
4.3 Synthesis of pyrazoles 2 and 3
Hydrazine monohydrate (390 µL, 7.8 mmol) was added to a solution of chrysin (100 mg, 0.393 mmol) in absolute ethanol (7 mL), and the mixture was stirred for 20 h under reflux. After that, the reaction solution volume was reduced approximately to 1/3 under reduced pressure, water was added (7 mL), and the resulting solution was extracted with DCM (3 × 7 mL). The DCM fractions were dried (anh. Na2SO4), filtered, and evaporated at reduced pressure. The obtained crude (87.0 mg, 0.34 mmoles) was dissolved in ethanol (7 mL), Selectfluor® was added (132.49 mg, 0.37 mmol) and the solution was stirred at room temperature for 20 h. Water was added (7 mL) and the aqueous solution was extracted with DCM (2 × 7 mL). The DCM fractions were dried (anh. Na2SO4), filtered, and evaporated at reduced pressure. The mixture was purified by MPLC-UV on reversed-phase silica gel using methanol/H2O gradients to obtain pyrazole 2 (22.8 mg, 28.5% yield) and pyrazole 3 (12.2 mg, 15.2% yield).
Pyrazole 2. Mp: 168–170 ℃. 1H NMR (300 MHz, (CD3)2CO): δ = 7.83–7.79 (m, 2H, Ar–H); 7.49–7.44 (m, 3H, Ar–H and Pyr-H); 7.39–7.37 (m, 1H, Ar–H); 6.96 (t, 1H, J = 8.10 Hz, Ar–H); 6.46 (d, 2H, J = 8.10 Hz, Ar–H). 13C NMR (75 MHz, (CD3)2CO): δ C1 = C3 = 157.99; C3' = 150.57; C5' = 143.70; C1'' = 130.45; C3'' = C5'' = 129.87; C4'' = 129.31; C5 = 129.27; C2'' = C6'' = 126.35; C4 = C6 = 107.99; C2 = 106.10; C4' = 104.76. IR (neat) ν = 3401, 1701, 1626, 1454, 1015, 768, 694 cm−1. HRMS: found m/z = 275.0783, calculated m/z for C15H12N2O2Na [M + Na]+ = 275.0791 (0.8 mDa error).
Pyrazole 3. Mp: 133–134 ℃. 1H NMR (300 MHz, (CD3)2CO: δ = 10.65 (1H, HO-Ar); 10.12 (1H, HO-Ar); 7.86 (m, 2H, Ar–H); 7.55 (s, 1H, Pyr-H); 7.52 (m, 2H, Ar–H); 7.43 (m, 1H, Ar–H); 6.94 (dd, 1H, J1 = 8.26 Hz, J2 = 4.08 Hz, Ar–H); 6.44 (dd, 1H, JHH = 8.9 Hz, JHF = 10.5 Hz, H-Ar). 13C NMR (75 MHz, (CD3)2CO: δ C1 = 153.51; C3' = 150.40; C4 = 146.53 (d, 1JC-F = 228.89 Hz); C3 = 145.40 (d, 2JC-F = 15.46 Hz); C5' = 144.00; C1'' = 130.31; C3'' = C5'' = 130.10; C4'' = 129.65; C2'' = C6'' = 126. 61; C5 = 115.35 (d, 2JC-F = 19.66 Hz); C2 = 107.88 (d, 3JC-F = 1.90 Hz); C6 = 106.48 (d, 3JC-F = 6.75 Hz); C4' = 104.92. 19F NMR (282 MHz, (CD3)2CO: δ = − 150.67 (1F, m). IR (neat) ν = 3415, 1703, 1634, 1494, 1462, 1026, 982, 854, 766, 695 cm−1. HRMS: found m/z = 271.0881, calculated m/z for C15H11FN2O2 [M + H]+ = 293.0877 (0.4 mDa error).
4.4 Microplate α-glucosidase inhibition assays
The hydrolysis of p-nitrophenyl-α-O-D-glucopyranoside (α-pNPG) was continuously measured in a 96-well microplate using a method similar to that applied by Arnaldos et al. [57]. Wells were filled in triplicate with α-Glc (yeast) in 0.1 M, pH 7 phosphate buffer (0.088 U/mL end concentration per well), α-cyclodextrin, the same buffer solution (1.22 mM end concentration per well) and 10 µL of test compound in dimethylsulfoxide (DMSO) solution. Wells containing the corresponding volume of DMSO without an inhibitor were used as the references of maximum enzymatic rates. The final volume per well was 270 µL. The enzymatic reaction was initiated by adding α-pNPG (1.63 mM end concentration per well). The plate was shaken for 2 s and the increase in absorbance at 405 nm was monitored at 37 ℃ for 10 min.
DMSO solutions (0.084 mg/mL) of doubly chemically modified extracts were employed for α-Glc inhibition percentage determination.
For IC50 determination, ten serial dilutions of the compounds were prepared in DMSO, following equally spaced points on a neperian logarithm scale, starting at 64.7 mM and finishing at 0.00647 mM (end concentration per well: 2397 to 0.2514 µM). IC50 calculated using Prism V5.01 (GraphPad Software Inc., La Jolla, CA, USA) applying a non linear regression curve fit for a log[inhibitor] vs. normalized answer model with variable slope. Standard drug acarbose was used as enzyme inhibition control.
4.5 TLC α-glucosidase inhibition assays
The α-Glc inhibition properties of the mixtures were surveyed by TLC autography using reported protocols [47]. Briefly, a Silica gel-TLC plate (64 cm2) was sprayed with the 2-naphthyl-α-D-glucopyranoside: Fast Blue B salt (1:1, V/V) solution using a glass reagent sprayer operated with compressed air. Then, the plate was dried under air current at room temperature. 80 mg of agar was dissolved at 80 ℃ in 9.4 mL phosphate buffer (100 mM, pH 7.0), the solution was allowed to cool down (40 ℃) and 188 μL of α-Glc solution (12.5 U/mL) was added. The obtained solution was mixed by inversion and distributed over the TLC plate. After cooling and solidification, the plate was incubated at 37 ℃ (10 min) in a stove.
4.6 HPLC conditions
DAD-HPLC measurements were performed on a Hewlett Packard HP 1050 series, coupled to a G1306AX DAD. The samples were directly loaded on a Phenomenex Gemini C18 column (5 µm × 150 mm × 4.6 mm). Mobile phase: A: Methanol with formic acid (5%); B water/methanol (39/61) with formic acid (5%). Method: 0 min, B 100%, 3 min B 100%, 5 min B 90%, 8 min B 90%, 15 min B 82%, 16 min B 82%, 17 min B 70%, 20 min B 60%, 22 min B 60%, 23 min B 10%, 25 min B 10%. Injected volume: 3 µl. Flow: 0.3 mL/min. Column temperature: 30.0 ℃. Detection at 254 nm. Solutions of 0.15 mg/mL (pure compounds) or 10 mg/mL (for mixture) were injected.
Notes
Acknowledgements
The authors thank Dr. German Rosano and the Instituto de Biología Molecular y Celular de Rosario (IBR, CONICET-UNR) for HRMS experiments, and the Instituto de Química de Rosario (IQUIR, CONICET-UNR) for the NMR experiments. M. I. Osella thanks the CONICET for her Doctoral Fellowship. M. O. Salazar and R. L. E. Furlan are CONICET Researchers.
Author contributions
María I. Osella: Investigation; Data curation; Formal analysis; Writing—review & editing. Mario O. Salazar: Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Supervision; Writing—original draft; and Writing—review & editing. Carlos M. Solís: Investigation; Writing—review & editing. Ricardo L.E. Furlan: Conceptualization; Methodology; Writing—original draft; Writing—review & editing; Supervision; Project administration; and Funding acquisition.
Availability of data and materials
The data supporting the results of this study can be obtained from the corresponding authors upon reasonable request.
Declarations
Competing interests
The authors declare that they have no conflict of interest.
References
-
1.Beato A, Haudecoeur R, Boucherle B, Peuchmaur M. Expanding chemical frontiers: approaches for generating diverse and bioactive natural product-like compounds libraries from extracts. Chem Eur J 2022;28: e202304166. CrossRef PubMed Google Scholar
-
2.Ramallo IA, Salazar MO, García P, Furlan RLE. Chemical diversification of natural product extracts. In: Rahman A, editor. Studies in natural products chemistry, vol. 60. Amsterdam: Elsevier; 2018. p. 371–98. PubMed Google Scholar
-
3.Li G, Lou H-X. Strategies to diversify natural products for drug discovery. Med Res Rev 2018;38: 1255-94. CrossRef PubMed Google Scholar
-
4.Ramallo IA, Salazar MO, Mendez L, Furlan RLE. Chemically engineered extracts: source of bioactive compounds. Acc Chem Res 2011;44: 241-50. CrossRef PubMed Google Scholar
-
5.Lopez SN, Ramallo IA, Gonzalez Sierra M, Zacchino SA, Furlan RLE. Chemically engineered extracts as an alternative source of bioactive natural product-like compounds. Proc Natl Acad Sci USA 2007;104: 441-4. CrossRef PubMed Google Scholar
-
6.Righi D, Marcourt L, Koval A, Ducret V, Pellissier L, Mainetti A, Katanaev VL, Perron K, Wolfender J-L, Ferreira QE. Chemo-diversification of plant extracts using a generic bromination reaction and monitoring by metabolite profiling. ACS Comb Sci 2019;21: 171-82. CrossRef PubMed Google Scholar
-
7.García P, Salazar MO, Ramallo IA, Furlan RLE. A new fluorinated tyrosinase inhibitor from a chemically engineered essential oil. ACS Comb Sci 2016;18: 283-6. CrossRef PubMed Google Scholar
-
8.García P, Ramallo IA, Salazar MO, Furlan RLE. Chemical diversification of essential oils, evaluation of complex mixtures and identification of a xanthine oxidase inhibitor. RSC Adv 2016;6: 57245-52. CrossRef PubMed Google Scholar
-
9.Mendez L, Salazar MO, Ramallo IA, Furlan RLE. Brominated extracts as source of bioactive compounds. ACS Comb Sci 2011;13: 200-4. CrossRef PubMed Google Scholar
-
10.Ramallo IA, Alonso VL, Rua F, Serra E, Furlan RLE. A bioactive trypanosoma cruzi bromodomain inhibitor from chemically engineered extracts. ACS Comb Sci 2018;20: 220-8. CrossRef PubMed Google Scholar
-
11.Solís CM, Salazar MO, Ramallo IA, García P, Furlan RLE. A tyrosinase inhibitor from a nitrogen-enriched chemically engineered extract. ACS Comb Sci 2019;21: 622-7. CrossRef PubMed Google Scholar
-
12.Salazar MO, Ramallo IA, Micheloni O, Gonzalez Sierra M, Furlan RLE. Chemically engineered extracts: bioactivity alteration through sulfonylation. Bioorg Med Chem Lett 2009;19: 5067-70. CrossRef PubMed Google Scholar
-
13.Salazar MO, Micheloni O, Escalante AM, Furlan RLE. Discovery of a β-glucosidase inhibitor from a chemically engineered extract prepared through sulfonylation. Mol Divers 2011;15: 713-9. CrossRef PubMed Google Scholar
-
14.Salazar MO, Osella MI, Ramallo IA, Furlan RLE. Nα-arylsulfonyl histamines as selective β-glucosidase inhibitors. RSC Adv 2018;8: 36209-18. CrossRef PubMed Google Scholar
-
15.Salazar MO, Osella MI, Arcusin DEJ, Lescano LE, Furlán RLE. New α-glucosidase inhibitors from a chemically engineered essential oil of Origanum vulgare L. Ind Crops Prod 2020;156: 112855. CrossRef PubMed Google Scholar
-
16.Tomohara K, Ohashi N, Uchida T, Nose T. Synthesis of natural product hybrids by the Ugi reaction in complex media containing plant extracts. Sci Rep 2022;12: 15568. CrossRef PubMed Google Scholar
-
17.Tomohara K, Maneenet J, Ohashi N, Nose T, Fujii R, Kim MJ, Sun S, Awale S. Ugi adducts as novel anti-austerity agents against PANC-1 human pancreatic cancer cell line: a rapid synthetic approach. Biol Pharm Bull 2023;46: 1412-20. CrossRef PubMed Google Scholar
-
18.Tomohara K, Ito T, Hasegawa N, Kato A, Adachi I. Direct chemical derivatization of natural plant extract: straightforward synthesis of natural plant-like hydantoin. Tetrahedron Lett 2016;57: 924-7. CrossRef PubMed Google Scholar
-
19.Wu T, Jiang C, Wang L, Morris-Natschke SL, Miao H, Gu L, Xu J, Lee KH, Gu Q. 3, 5-Diarylpyrazole derivatives obtained by ammonolysis of the total flavonoids from chrysanthemum indicum extract show potential for the treatment of Alzheimer's disease. J Nat Prod 2015;78: 1593-9. CrossRef PubMed Google Scholar
-
20.Zhang J-L, Xu W, Zhou Z-R, Li J, Jiang L-L, Zhang X-X, Jiang R-W. Antineoplastic constituents from the chemical diversified extract of radix puerariae. Chem Biodiversity 2019;16: e1800408. CrossRef PubMed Google Scholar
-
21.Kikuchi H, Sakurai K, Oshima Y. Development of diversity-enhanced extracts of curcuma zedoaria and their new sesquiterpene-like compounds. Org Lett 2014;16: 1916-9. CrossRef PubMed Google Scholar
-
22.Suzuki Y, Ichinohe K, Sugawara A, Kida S, Zhang J, Yamada O, Hattori T, Oshima Y, Murase S, Kikuchi H. Development of indole alkaloid-type dual immune checkpoint inhibitors against CTLA-4 and PD-L1 based on diversity-enhanced extracts. Front Chem 2021;9: 766107. CrossRef PubMed Google Scholar
-
23.Kikuchi H, Kawai K, Nakashiro Y, Yonezawa T, Kawaji K, Kodama EN, Oshima Y. Construction of a meroterpenoid-like compounds library based on diversity-enhanced extracts. Chem Eur J 2019;25: 1106-12. CrossRef PubMed Google Scholar
-
24.Du Y, Sun J, Gong Q, Wang Y, Fu P, Zhu W. New α-pyridones with quorum-sensing inhibitory activity from diversity-enhanced extracts of a Streptomyces sp. derived from marine algae. J Agric Food Chem 2018;66: 1807-12. CrossRef PubMed Google Scholar
-
25.Giri GF, Viarengo G, Furlan RLE, Suárez AG, Garcia Véscovi E, Spanevello RA. Soybean hulls, an alternative source of bioactive compounds: combining pyrolysis with bioguided fractionation. Ind Crops Prod 2017;105: 113-23. CrossRef PubMed Google Scholar
-
26.Adessi TG, Ana Y, Stempin CC, García MC, Bisogno FR, Nicotra VE, García ME. Psilostachyins as trypanocidal compounds: bioguided fractionation of Ambrosia tenuifolia chemically modified extract. Phytochem 2022;194: 113014. CrossRef PubMed Google Scholar
-
27.Tan Y, Sun X, Dong F, Tian H, Jiang R. Enhancing the structural diversity and bioactivity of natural products by combinatorial modification exemplified by total tanshinones. Chin J Chem 2015;33: 1084-8. CrossRef PubMed Google Scholar
-
28.Kamauchi H, Noji M, Kinoshita K, Takanami T, Koyama K. Coumarins with an unprecedented tetracyclic skeleton and coumarin dimers from chemically engineered extracts of a marine-derived fungus. Tetrahedron 2018;74: 2846-56. CrossRef PubMed Google Scholar
-
29.Papoutsis K, Zhang J, Bowyer MC, Brunton N, Gibney ER, Lyng J. Fruit, vegetables, and mushrooms for the preparation of extracts with α-amylase and α-glucosidase inhibition properties: a review. Food Chem 2021;338: 128119. CrossRef PubMed Google Scholar
-
30.Nyenwe EA, Jerkins TW, Umpierrez GE, Kitabchi AE. Management of type 2 diabetes: evolving strategies for the treatment of patients with type 2 diabetes. Metabolism 2011;60: 1-23. CrossRef PubMed Google Scholar
-
31.Hu C, Jia W. Therapeutic medications against diabetes: what we have and what we expect. Adv Drug Deliv Rev 2019;139: 3-15. CrossRef PubMed Google Scholar
-
32.Dai T, Chen J, McClements DJ, Li T, Liu T. Investigation the interaction between procyanidin dimer and α-glucosidase: spectroscopic analyses and molecular docking simulation. Int J Biol Macromol 2019;130: 315-22. CrossRef PubMed Google Scholar
-
33.Narender T, Madhur G, Jaiswal N, Agrawal M, Maurya CK, Rahuja N, Srivastava AK, Tamrakar AK. Synthesis of novel triterpene and N-allylated/N-alkylated niacin hybrids as α-glucosidase inhibitors. Eur J Med Chem 2013;63: 162-9. CrossRef PubMed Google Scholar
-
34.Nguyen DV, Shaw LC, Grant MB. Inflammation in the pathogenesis of microvascular complications in diabetes. Front Endocrinol 2012;3: 170. CrossRef PubMed Google Scholar
-
35.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature 2001;414: 782-7. CrossRef PubMed Google Scholar
-
36.Raut JS, Karuppayil SM. A status review on the medicinal properties of essential oils. Ind Crops Prod 2014;62: 250-64. CrossRef PubMed Google Scholar
-
37.Maffei ME, Gertsch J, Appendino G. Plant volatiles: production, function and pharmacology. Nat Prod Rep 2011;28: 1359-80. CrossRef PubMed Google Scholar
-
38.Lescano LE, Salazar MO, Furlan RLE. Chemically engineered essential oils prepared through thiocyanation under solvent-free conditions: chemical and bioactivity alteration. Nat Prod Bioprospect 2024;14: 35. CrossRef PubMed Google Scholar
-
39.Lazović M, Ivković Đ, Jankov M, Dimkić I, Janakiev T, Trifković J, Milojković-Opsenica D, Ristivojević P. Enhancement of propolis food preservation and functional ingredient characteristics by natural eutectic solvents extraction of phytochemicals. Food Biosci 2024;57: 103467. CrossRef PubMed Google Scholar
-
40.Tran TD, Ogbourne SM, Brooks PR, Sánchez-Cruz N, Medina-Franco JL, Quinn RJ. Lessons from exploring chemical space and chemical diversity of propolis components. Int J Mol Sci 2020;21: 4988. CrossRef PubMed Google Scholar
-
41.Santos LM, Fonseca MS, Sokolonski AR, Deegan KR, Araújo RP, Umsza-Guez MA, Barbosa JD, Portela RD, Machado BA. Propolis: types, composition, biological activities, and veterinary product patent prospecting. J Sci Food Agric 2020;100: 1369-82. CrossRef PubMed Google Scholar
-
42.Huang S, Zhang CP, Wang K, Li GQ, Hu FL. Recent advances in the chemical composition of propolis. Molecules 2014;19: 19610-32. CrossRef PubMed Google Scholar
-
43.Tsuda T, Kumazawa S. Propolis: chemical constituents, plant origin, and possible role in the prevention and treatment of obesity and diabetes. J Agr Food Chem 2021;69: 15484-94. CrossRef PubMed Google Scholar
-
44.Bohmdorfer S. Effect-directed detection: chemical and enzyme-based assays. In: Poole CF, editor. Instrumental thin-layer chromatography. 2nd ed. Amsterdam: Elsevier; 2023. p. 297–324. PubMed Google Scholar
-
45.Cabezudo I, Salazar MO, Ramallo IA, Furlán RLE. Effect-directed analysis in food by thin-layer chromatography assays. Food Chem 2022;390: 132937. CrossRef PubMed Google Scholar
-
46.Legerská B, Chmelová D, Ondrejovič M, Miertuš S. The TLC-bioautography as a tool for rapid enzyme inhibitors detection—a review. Crit Rev Anal Chem 2022;52: 275-93. CrossRef PubMed Google Scholar
-
47.Capozza GP, Salazar MO, Ramallo IA, Furlan RLE. Development of a thin-layer chromatography gel-overlay α-glucosidase inhibition assay. JPC J Planar Chromat 2023;36: 483-91. CrossRef PubMed Google Scholar
-
48.Solis CM, Salazar MO, Ramallo IA, García P, Furlan RLE. Cyclocondensation versus cyclocondensation plus dehydroxylation during the reaction of flavones and hydrazine. Eur J Org Chem 2022;25: e202200455. CrossRef PubMed Google Scholar
-
49.Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 2017;7: 42717. CrossRef PubMed Google Scholar
-
50.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2001;46: 3-26. CrossRef PubMed Google Scholar
-
51.Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem 1999;1: 55-68. CrossRef PubMed Google Scholar
-
52.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 2002;45: 2615-23. CrossRef PubMed Google Scholar
-
53.Egan WJ, Merz KM Jr, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem 2000;43: 3867-77. CrossRef PubMed Google Scholar
-
54.Muegge I, Heald SL, Brittelli D. Simple selection criteria for drug-like chemical matter. J Med Chem 2001;44: 1841-6. CrossRef PubMed Google Scholar
-
55.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 2010;53: 2719-40. CrossRef PubMed Google Scholar
-
56.Brenk R, Schipani A, James D, Krasowski A, Gilbert IH, Frearson J, Wyatt PG. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem 2008;3: 435-44. CrossRef PubMed Google Scholar
-
57.Arnaldos TL, Serrano ML, Calderón AA, Muñoz R. A rapid and continuous spectrophotometric method to measure β-glucosidase activity based on p-nitrophenyl β-O-D-glucopyranoside hydrolysis. Phytochem Anal 1999;10: 171-4. CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2025
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
