


Antiviral and anti-inflammatory activities of chemical constituents from twigs of Mosla chinensis Maxim
This paper was financed by National Natural Science Foundation of China (No. 31660100), Innovative Team of Yunnan Province (No. 2019HC018), the key Project of Yunnan Province (No. 202103AC10005, No. 202302AG050004)
Abstract
Seven undescribed compounds, including three flavones (1–3), one phenylpropanoid (19), three monoaromatic hydrocarbons (27–29), were isolated from the twigs of Mosla chinensis Maxim together with twenty-eight known compounds. The structures were characterized by HRESIMS, 1D and 2D NMR, and ECD spectroscopic techniques. Compound 20 displayed the most significant activity against A/WSN/33/2009 (H1N1) virus (IC50 = 20.47 μM) compared to the positive control oseltamivir (IC50 = 6.85 µM). Further research on the anti-influenza mechanism showed that compound 20 could bind to H1N1 virus surface antigen HA1 and inhibit the early attachment stage of the virus. Furthermore, compounds 9, 22, 23, and 25 displayed moderate inhibitory effects on the NO expression in LPS inducing Raw 264.7 cells with IC50 values of 22.78, 20.47, 27.66, and 30.14 µM, respectively.Graphical Abstract
Keywords
Mosla chinensis Maxim Flavonoids Phenolic structure Anti-H1N1 virus activity Anti-inflammatory activity1 Introduction
Influenza viruses had high pathogenicity and infectiousness, and is an important risk factor for human health. It had been exhibited the ability to invade the epithelial cells of the respiratory tract for the happening of the inflammation, and thereby result in influenza with the symptom such as fever, headache, and muscle pain. Influenza is one of the most common respiratory diseases. If the patients had not effective medical interventions, it could induce serious complications such as pneumonia, acute lung injury and even pulmonary fibrosis [1, 2]. Influenza viruses induced diseases had been become a worldwide public health problem and the main treatment is vaccine or drug. However, because of the extraordinary high rate of virus mutation and the side effects of existing drugs, it's essential to find ingredients with high effect and low toxicity from natural food. Phenolic compounds containing multiple phenolic hydroxyl groups, which can bind with targeting proteins of disease and possess significant activities of antioxidant, antiviral, and anti-inflammatory.
Mosla chinensis Maxim, recorded in Chinese Pharmacopoeia, is a medicinal and edible plant, which mainly distributed in southern China [3]. It belongs to the Labiatae family, a tomentose and aromatic plant that traditionally has been used as an herbal drug to treat colds in wet summers and aversion to cold with fever [4]. The leaves of M. chinensis are widely used as vegetable, herbal tea, beverage or food additives because of its human beneficial properties in China. Furthermore, M. chinensis is a productive source of essential oil and flavonoids. Several investigations have shown that essential oil of M. chinensis have the activities of antioxidant and antimicrobial [5-9] and the flavonoids exhibited the activities of anti-influenza A virus [10]. In our continuous search for compound of anti-influenza virus [11, 12], we found that few studies on the antiviral activity of other compounds isolated from M. chinensis were carried out. So, this paper was focus on exploring the activity of phenolic compounds. In this study, we investigated the extraction, structural analysis, biological activities and their possible mechanism researches of 35 compounds (covering 7 undescribed compounds and 28 known compounds) in the M. chinensis twigs.
2 Results and discussion
2.1 Structure characterization of the isolated compounds from M. chinensis
A comprehensive phytochemical investigation resulted in the isolation and identification of 35 compounds including seven new compounds (1–3, 19, and 27–29) and twenty-eight known compounds. The known compounds were listed as follows: 8-(4ʹʹ-hydroxyphenyl)-5,7,4ʹ-trihydroxyflavone (4) [13], apigenin-7-O-glucuronide methyl ester (5) [14], acacetin-7-O-glucuronide methyl ester (6) [15], Isolinariin B (7) [16], acacetin 7-O-[6ʹʹʹ-O-acetyl-β-D-galactopyranosyl-(1 → 3)]-β-D-xylopyranoside (8) [17], luteolin (9) [18], apigenin 7-O-β-D-glucopyranoside (10) [19], apigenin 4ʹ-O-β-D-glucopyranoside (11) [19], acacetin 7-O-β-D-xylopyranoside (12) [20], 4ʹ,5,7-trihydroxy-3ʹ,5ʹ-dimethoxyflavone 7-O-[β-D-apiofuranosyl (1ʹʹʹ → 2ʹʹ)]-β-D-glucopyranoside (13) [21], acacetin 7-O-β-D-apiofuranosyl-(1ʹʹʹ → 2ʹʹ)-β-D-glucopyranoside (14) [22], Diosmetin 7-O-β-D-xylopyranoside (15) [23], acacetin 7-O-[4ʹʹʹ-O-acetyl-β-D-apiofuransyl-(1ʹʹʹ → 3ʹʹ)]-β-D-xylopyranoside (16) [24], Sakuranetin (17) [25], Pyrroside A (18) [26], methyl lithospermate (20) [27], dimethy lithospermate (21) [27], hyprhombins A (22) [28], 3-(3ʹʹʹ, 4ʹʹʹ-dihydroxyphenyl)-acrylic acid 1-(3ʹʹ, 4ʹʹ-dihydroxyphenyl)-2-methoxycarbonylethyl ester (23) [29], sebestenoids C (24) [30], methyl salvianol acid C (25) [31], agrimonolide 6-O-β-D-glucopyranoside (26) [32], 3ʹ-hydroxyphenyl-3,4,5-trimethylgallate (30) [33], 4-[[(4-hydroxybenzoyl)oxy]methyl]phenyl-β-D-glucopyranoside (31) [34], 4-O-β-D-glucopyranosylbenzyl-3ʹ-hydroxyl-4ʹ-methoxybenzoate (32) [34], 4-[[(2ʹ, 5ʹ-dihydroxybenzoyl)oxy]methyl]phenyl-O-β-D-glucopyranoside (33) [19], amburoside A (34) [35], 4-hydroxybenzyl alcohol 4-O-[5-O-(4-hydroxy)benzoyl]-β-D-apiofuranosyl (1 → 2)-β-D-glucopyranoside (35) [36].
Compound 1, yellow amorphous powder, gave the molecular formula of C26H28O13 based on its HR-ESI–MS ([M + Na] + m/z 571.1408, calcd. 571.1428). The 1H NMR spectroscopic data (Table 1) of 1 showed the signals for six aromatic protons at δH 8.09 (2H, d, J = 8.6 Hz, H-2ʹ/6ʹ), 7.14 (2H, d, J = 8.6 Hz, H-3ʹ/5ʹ), 6.83 (1H, s, H-8), 6.41 (1H, s, H-6), an olefinic proton at δH 6.97 (1H, s, H-3), a hydroxyl at δH 12.96 (1H, s), and one methyl group at δH 3.87 (3H, s, H-OMe). Two anomeric protons δH 5.35 (1H, d, J = 6.7 Hz, H-1ʹʹʹ), 5.18 (1H, d, J = 7.5 Hz, H-1ʹʹ) were observed, which imply the presence of two aglycons. Acid hydrolysis afforded two sugar components as detected by the coupling constant values (JH-1ʹʹ, H-2ʹʹ) and the GC analysis as β-D-xylose and β-D-apiose (Additional file 1: Fig. S60). The 13C NMR spectrum of 1 (Table 1) showed characteristic signals for the flavonoid skeleton at δC 182.5 (C-4), 164.2 (C-2), 99.8 (C-6), and 95.0 (C-8). The pyranose from of the sugars was also revealed from the 13C-NMR chemical shift values (Table 1) [37-39]. Based on 1H, 13C NMR and HSQC data, the signals at δC 163.2 (C-4ʹ), 162.9 (C-7), 161.3 (C-5), δH 6.83 (1H, s, H-8), and 6.41 (1H, s, H-6) shows that 1 is an 5,7,4ʹ-trisubstituted flavonoid.
1H (600 MHz) and 13C (150 MHz) NMR data of compounds 1–3 (δ in ppm, J in Hz)
The 1H and 13C NMR spectroscopic data of 1 (Table 1) is highly similar to acacetin [40], except for the additional presence of sugar moiety in 1. The above observation indicated 1 was glycoside derivative of acacetin. The position of glycosyl junction was identified by HMBC map. A series of HMBC correlations from HXyl-1 (δH 5.18) to C-7 (δC 162.9), from HApi-1 (δH 5.35) to CXyl-4 (δC 69.9), from HXyl-4 (δH 3.43) to CApi-1 (δC 109.3), enable the sugar chain of C-7 to be assigned as 7-O-[β-D-apiofuransyl-(1ʹʹʹ → 4ʹʹ)]-β-D-xylopyranoside. Thus, the structure of 1 was elucidated as acacetin 7-O-[β-D-apiofuransyl-(1ʹʹʹ → 4ʹʹ)]-β-D-xylopyranoside.
Compound 2 was purified as a yellow amorphous powder with the molecular formula of C31H34O16 according to the HR-ESI–MS spectrum ([M + H] + m/z 663.1910, calcd. 663.1920). The 1H and 13C NMR spectroscopic data of 2 (Table 1) were highly analogue to those of 1 except the presence of two acetyl groups at δC 170.5, 170.7, 20.7, and 21.1 in 2, as well as the minor change of chemical shifts in two sugars. Hence, 2 was deduced to be the acylated derivative of 1. Acid hydrolysis demonstrated the glycosidic nature of 2, which was identified as the β-D-glucose and β-D-apiose by the GC analysis and coupling constant values (JH-1ʹʹ, H-2ʹʹ) (Additional file 1: Fig. S61). The position of glycosyl junction was identified by the HMBC correlations from HGlc-1 (δH 5.25) to C-7 (δC 163.0), from HApi-1 (δH 5.29) to CGlc-2 (δC 76.9), from HGlc-2 (δH 4.04) to CApi-1 (δC 108.5). Furthermore, the sequence of the acetyl groups was deduced to be connected to CGlc-6 and CApi-5 due to the HMBC correlations from HGlc-6 (δH 3.79/3.20) to C-7ʹʹ (δC 170.5), from HApi-5 (δH 3.66/3.52) to C-6ʹʹʹ (δC 170.7). Therefore, compound 2 was identified as acacetin 7-O-[5ʹʹʹ-O-acetyl-β-D-apiofuransyl-(1ʹʹʹ → 2ʹʹ)]-6ʹʹ-O-acetyl-β-D-glucoside (Fig. 1).

Structures of compounds 1–35 isolated from M. chinensis Maxim
Compound 3 was obtained as a yellow amorphous powder. It showed a quasi-molecular ion peak at m/z 579.1717 [M + H] + (calcd. 579.1714) in the HR-ESI–MS data, suggesting a molecular formula C27H30O14. The 1H and 13C NMR spectroscopic data of 3 (Table 1) were also very similar to those of 1, apart from the extra methoxy group at C-3ʹ in 3 rather than hydrogen group at C-3ʹ in 1, which was supported by HMBC correlation between H-3ʹ-OMe (δH 3.67, s) and C-3ʹ (147.6) (Fig. 2). The anomeric configuration of D-xylose and D-apiose was confirmed to be β-configuration, according to the J value (J = 7.5 and 6.7 Hz) of the anomeric proton in the two sugar units (Additional file 1: Fig. S62). Thus, the structure of 3 was elucidated to be 3ʹ, 4ʹ-dimethoxyluteolin-7-O-[β-D-apiofuransyl-(1ʹʹʹ → 4ʹʹ)]-β-D-xylopyranoside.
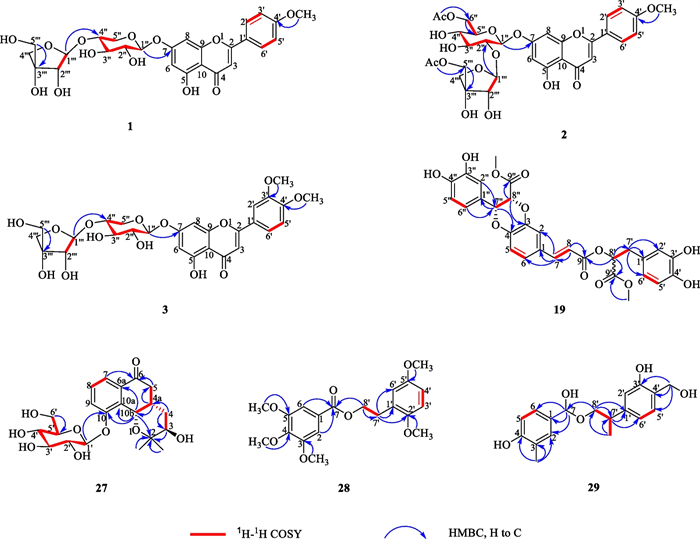
1H-1H COSY and key HMBC correlations of compounds 1–3, 19 and 27–29
Compound 19, white amorphous powder, gave molecular formula of C29H26O12 deduce from the HRESIMS spectrum (m/z 589.1293 [M + Na] +, calcd. 589.1322). The 1H NMR spectroscopic data (Table 2) of 19 showed signals for two olefinic methine protons at δH 7.58 (1H, d, J = 16.0 Hz, H-7), 6.37 (1H, d, J = 16.0 Hz, H-8), three oxymethine protons at δH 5.19 (1H, dd, J = 8.0, 5.0 Hz, H-8ʹ), 5.14 (1H, d, J = 5.0 Hz, H-7ʹʹ), and 5.07 (1H, d, J = 5.0 Hz, H-8ʹʹ), two methoxy protons at δH 3.69 (3H, s, H-9ʹʹ-OMe) and 3.62 (3H, s, H-9ʹ-OMe), two methylene protons at δH 3.03 (1H, dd, J = 13.5, 8.0 Hz)/3.31 (1H, dd, J = 13.5, 5.8 Hz), and nine phenyl protons. The spectra of 13C NMR and DEPT displayed twenty-nine carbon signals, covering twelve quaternary carbons [containing three carbonyls at δC 170.7 (C-9ʹʹ), 168.1 (C-9ʹ), 166.5 (C-9)], fourteen methines (including three oxymethines at δC 76.6, 75.8, and 73.4, two olefinic carbons at δC 145.9 and 116.1, and nine phenyl carbons), one methylene, and two methyls. A comprehensive analysis of the 1H and 13C NMR spectral data (Table 2) revealed that 19 was a lignin, structurally comparable to clinopodic acid C [41] except for the lack of signals for two methoxy groups at C-9ʹ and C-9ʹʹ in clinopodic acid C (Fig. 2). The two methoxy groups were assumed to be connected to C-9ʹ and C-9ʹʹ, respectively, according to the HMBC correlations of H-9ʹʹ-OMe (δH 3.69) with C-9ʹʹ (δC 170.7) and H-9ʹ-OMe (δH 3.62) with C-9ʹ (δC 168.1). According to biogenetic considerations and key ROESY correlations (Fig. 3) observed between H-7ʹʹ/H-8ʹʹ, H-7ʹʹ/H-6ʹʹ and H-7ʹʹ/H-2ʹʹ suggested that H-7ʹʹ and H-8ʹʹ were located on the same side of the ring. The CD spectrum showed a negative Cotton effect at 244 nm (Fig. 4), suggesting that the absolute configuration of the benzodioxane moiety was established as 7ʹʹR, 8ʹʹR [41]. Therefore, compound 19 is identified as 9ʹ, 9ʹʹ-dimethyl clinopodic acid C.
1H (600 MHz) and 13C (150 MHz) NMR data of compounds 19, 27–29 (in ppm, J in Hz)
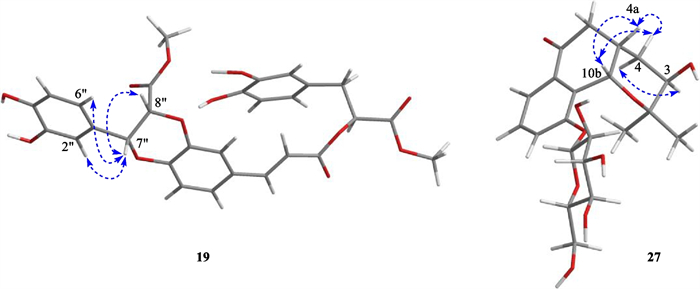
Key ROESY correlations of compounds 19 and 27
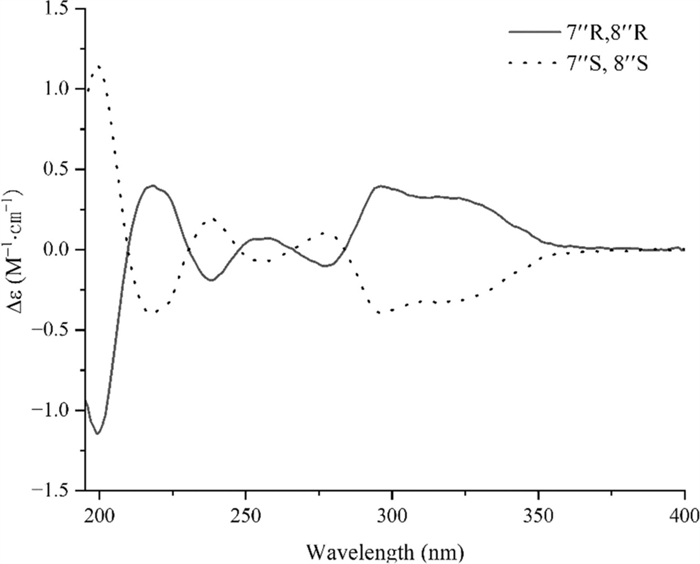
Experimental CD spectra of 19
Compound 27 was isolated as a colorless oil. The molecular formula of 27 was assigned as C21H28O9 based on the HR-ESI–MS at m/z 447.1638 [M + Na]+ (calcd. 447.1631). Analyses of the 1D NMR data (Table 2) of 27 revealed signals for one ketone (δC 199.7), one trisubstituted benzene ring [δC 157.8, 133.7 and 133.5; δH 7.67 (d), 7.54 (d) and 7.45 (t)], and two methyls [δC 25.9 and 24.6; δH 1.10 (s) and 1.04 (s)]. In addition, one anomeric proton at δH 5.01 (1H, d, J = 7.7 Hz, H-1ʹ) was observed, which implies the presence of one aglycon. Acid hydrolysis of 27 afforded one sugar moiety, which was identified as the β-D-glucose by the GC analysis and coupling constant values (JH-1ʹ, H-2ʹ) (Additional file 1: Fig. S63). The 1D NMR spectroscopic data of 27 (Table 2) were virtually identical to those of (3R, 4aR, 10bR)-3,10-dihydroxy-2,2-dimethyl-3,4,4a,10b-tetrahydro-2H-naphtho[1,2-b]-pyran-5H-6-one [42]. The major difference is the presence glycosyl group of located at C-10 in 27 as reinforced by the HMBC correlations from HGlc-1 (δH 5.01) to C-10 (δC 157.8).
The relative configuration of 27 was determined by ROESY correlations (Fig. 3) of H-10b/H-4a, H-4a/H-4β, H-10b/H-4β, H-4α/H-3 observed, determined that the junction of B/C ring adopted a cis configuration, suggesting H-10b and H-4a were located on the same side of the ring C, and H-3 was the opposite. Hence, two stereoisomers were conceivable at that point, namely 3R, 4aS, 10bS and 3S, 4aR, 10bR. Subsequently, the absolute configuration of 27 was assigned as 3R, 4aS, and 10bS by comparison of the calculated and experimental ECD data in Fig. 5. Therefore, compound 27 was identified as a (3R, 4aS, 10bS)-2,2-dimethyl-3-hydroxy-10-O-β-D-glucoside-3,4,4a,10b-tetrahydro-2H-naphtho[1,2-b]-pyran-5H-6-one, named Mosla chinensis glycoside B1.
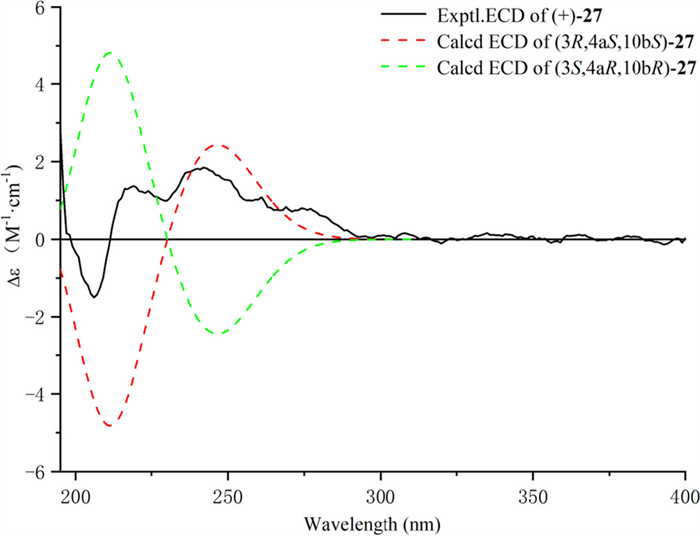
Calculated and experimental ECD spectra of 27; σ = 0.20 eV; UV shift = − 30 nm
Compound 28, white amorphous powder, possesses a molecular formula of C20H24O7 concluded form the HR-ESI–MS spectrum ([M + H] + m/z 377.1592, calcd. 377.1595). The 1H NMR spectrum (Table 2) of 28 revealed the signal for five aromatic protons at δH 7.25 (2H, s, H-2/6), 6.90 (1H, d, J = 8.2 Hz, H-3ʹ), 6.88 (1H, s, H-6ʹ), and 6.85 (1H, d, J = 8.2 Hz, H-4ʹ), two methylene groups at 4.48 (2H, t, J = 6.8 Hz, H-8ʹ) and 3.00 (2H, t, J = 6.8 Hz, H-7ʹ), and five methoxy protons at δH 3.85 (6H, s, H-3/5-OMe), 3.81 (3H, s, H-2ʹ-OMe), 3.79 (3H, s, H-4-OMe), and 3.78 (3H, s, H-5ʹ-OMe). Twenty carbon signals were totally observed in the spectra of 13C NMR and DEPT, covering eight quaternary carbons (including one ketone carbonyl at δC 167.5), five methyls, two methylenes (including one oxymethylene at δC 66.9), and five methines (Table 2). High similarity in the spectra of 1D NMR was found between 28 (Table 2) and 2-phenylethyl 2,4-dihydroxy-3-methylbenzoate [43]. The main difference between them is the presence of five methoxy groups at C-3, 4, 5, 2ʹ, 5ʹ in 28 rather than the two hydroxy groups at C-3, 5 and one methyl group at C-4 in 2-phenylethyl 2,4-dihydroxy-3-methylbenzoate. The five methoxy groups were assumed to be connected to C-3, 4, 5, 2ʹ, 5ʹ, respectively, according to the HMBC correlations of C-3-OMe (δH 3.85) with C-3 (δC 56.6), C-4-OMe (δH 3.79) with C-4 (δC 56.4), C-5-OMe (δH 3.85) with C-5 (δC 56.6), and C-2ʹ-OMe (δH 3.81) with C-2ʹ (δC 61.1). Therefore, the structure of 28 was assigned as 2ʹ,5ʹ-dimethoxyphenethyl 3,4,5-trimethoxybenzoate.
Compound 29, white amorphous powder, protonated a molecule peak at m/z 319.1537 [M + H] + (calcd 319.1540) corresponding to the molecular formula of C18H22O5. The 1H NMR spectrum of 29 exhibited six aromatic protons at δH 7.10 (2H, d, J = 8.1 Hz, H-6,5ʹ), 7.09 (2H, d, J = 8.1 Hz, H-5,6ʹ), 6.58 (1H, s, H-2ʹ), 6.33 (1H, s, H-2) and aliphatic protons at δH 5.50 (1H, s, H-7), 3.92 (1H, dd, J = 11.2, 3.7 Hz, H-8ʹa), 3.78 (1H, dd, J = 11.2, 2.9 Hz, H-8ʹb), 3.34 (2H, s, H-4ʹ-CH2), 2.57 (1H, qt, J = 7.1, 3.2 Hz, H-7ʹ), 2.00 (3H, s) and 1.38 (3H, d, J = 7.1 Hz). The 13C NMR spectrum of 29 exhibited 18 carbon signals, which including six quaternary carbons [covering three oxygen-bearing sp3 carbons at 79.4 (C-7), 69.1 (C-8ʹ), 62.9 (C-4ʹ-CH2)], eight methine, two methyl, and two methylene. Three 1H-1H COSY correlated systems of H-5/H-6; H-7ʹ/ H-8ʹ, and H-5ʹ /H-6ʹ were observed (Fig. 2). The results indicated that compound 29 was deduced to be a monoaromatic hydrocarbon and was structurally similar to that of 2-phenylethyl 2,4-dihydroxy-3-methylbenzoate [44]. The significant difference between them were substituent groups at C-2, 7, 3ʹ, 4ʹ, 7ʹ, there were hydrogen, hydroxy, hydroxy, hydroxymethyl and methyl at C-2, 7, 3ʹ, 4ʹ, 7ʹ of 29 rather than hydroxy, carbonyl, hydrogen, hydrogen and methyl. The deduction can be verified by the HMBC correlations of C-7ʹ (δH 2.75) with C-1ʹ (δC 138.8), C-7 (δH 5.50) with C-1 (δC 134.9), C-8ʹ (δH 3.92, 3.78) with C-7 (δC 79.4) and C-7ʹ (δC 31.6), C-7ʹ-Me (δH 1.38) with C-7ʹ (δC 36.1), C-4ʹ-CH2 (δH 3.34) with C-4ʹ (δC 127.4). Therefore, compound 29 was identified as 4-(hydroxy(2-(3-hydroxy-4-(hydroxymethyl) phenyl) propoxy) methyl)-2-methylphenol.
2.2 Biological evaluation
2.2.1 Anti-influenza A virus activity
The activity of compounds (6–18, 20–26, 31–35) against the influenza virus was evaluated by using A/WSN/33/2009 (H1N1) infected MDCK cells. In comparison with the positive control oseltamivir with IC50 = 6.85 µM, compound 20 exhibited significant inhibition effects of H1N1 (IC50 = 20.47 µM); However, other compounds had no anti-influenza activity. The results of western blot analysis showed that 20 could dramatically reduce the nucleoprotein protein expression at 2, 5, and 8 h, indicating that 20 inhibits influenza virus infection by interfering with the beginning phase in the viral life cycle (Fig. 6). Furthermore, the nucleoprotein distribution in infected cells was observed by fluorescence microscopy (Fig. 7). It showed that after virus infection for 2 and 5 h, the virus population in the MDCK cells of the DMSO group was dramatically higher than that of the experimental group. This result further indicated that the influenza virus could be inhibited by compound 20.
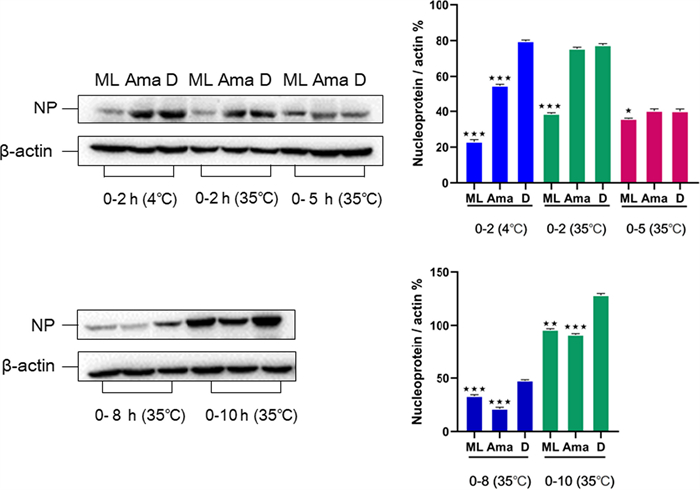
Effect of 20 (50 μM) on the expression of nucleoprotein in MDCK cells. Aam was amantadine, and D stand for DMSO control. Protein expression level and gray percentage of nucleoprotein and β-actin in 0–2 h (4 ℃), 0–2 h, 0–5 h, 0–8 h and 0–10 h (35 ℃). (*P < 0.05, **P < 0.01, ***P < 0.001 vs IAV group, using one-way ANOVA method)

Indirect immunofluorescence microscopy. MDCK cells were infected with A/WSN/33/2009 (H1N1) and treated with 20 (50 μM). After 2, 5, 8, and 10 h post infection, the cells were fixed for 30 min at 4 ℃ (A–D). Cell nuclei were stained with DAPI (blue) and viewed using a fluorescence microscopy (Magnification 400 ×)
Glycoprotein hemagglutinin (HA) of the influenza virus has been used as a potentially important target for developing anti-influenza drugs [45]. A hemagglutinin inhibition (HI) assay was designed to check if 20 could prevent virus attachment to the cells through disturbing the connection between HA and cellular receptors. The results showed that 20 might effectively promote erythrocyte agglutination at 40 μM (Fig. 8A and B). The results indicated that 20 could bind to influenza virus surface antigen HA1, inhibiting the early adsorption process of WSN. In addition, it was revealed that the docking sites with high-affinity underlying interaction could be intently related to residues ASN 68 and ARG 224 through Molecular docking (Fig. 8C). However, no binding sites between 20 and HA were observed on the receptor binding domain (RBD) of HA1 sialic acids. Therefore, it could be concluded that 20 exhibited the antiviral influence on A/WSN/33/2009 (H1N1) virus by targeting the hemagglutinin fusion machinery.
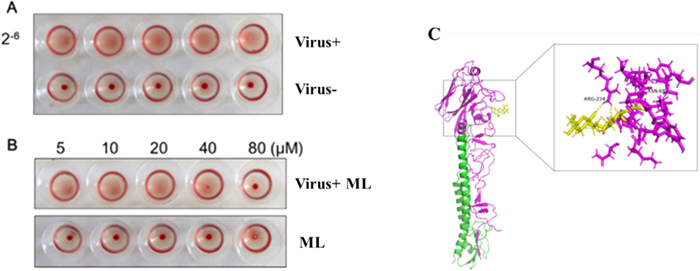
Effects of 20 on HA1. A The hemagglutination titer of WSN was 2–6, and red blood cells mixed with virus could not agglutinate. Normal red blood cells produce cell agglutination at room temperature. B The compound could effectively promote erythrocyte agglutination at 40 μΜ and 80 μΜ, the compound had no effect on red blood cells. C The HA1 polypeptide is colored purple, HA2 is green, and 20 is yellow. 20 can bind with HA1 residues
2.2.2 Anti-inflammatory activity
The influenza virus can lead to an excessive immune response and induce the production of inflammatory cytokines, such as IL-1 and IL-6 [46]. So, it would be valuable if the drugs had both antiviral and anti-inflammatory activities. Hence, we used the cells (LPS-activated RAW 264.7) to evaluate the impact of compounds (6–18, 20–26, 31–35) on preventing NO production. It was found that compounds 9, 22, 23, and 25 rendered moderate activity in preventing NO production (IC50 = 22.78, 20.47, 27.66, and 30.14 µM, respectively), in comparison with the positive control L-NMMA (IC50 = 21.80 µM) (Table 3).
Inhibitory effect of 9, 22, 23 and 25 on LSP-induced NO production in macrophages
3 Experimental procedures
3.1 General experimental procedures
A Jasco digital polarimeter (DIP-370, purchased from JASCO Corporation, Tokyo, Japan) was employed to examine optical rotations. NMR spectra were monitored by a Bruker AV 600 MHz spectrometer using an internal standard (tetramethylsilane) (Bruker BioSpin Group, Germany). An API-QSTAR Pulsar (Applied Biosystem Corporation, Canada) was hired to achieve HR-ESI–MS and ESI–MS. A Shimadzu UV-2401 spectrometer (Beckman, Brea, USA) was implemented to obtain the UV spectra. Column chromatography with various gels was conducted, including 75 μM of ODS-C18 (YMC Co., Ltd., Japan), 75–150 μM of MCI gel (GHP20P, Mitsubishi Chemical Corporation, Tokyo, Japan), 43–63 mm of LiChroprep RP-18 (Merck), 80–100 & 200–300 mesh of silica gels (Qingdao Marine Chemical Co., Ltd., China), and Sephadex LH-20 (Amersham Biosciences AB, Uppsala, Sweden). An Agilent 1260 liquid chromatography system (Agilent, USA) was implemented for semipreparative and analytical HPLC analysis on a semipreparative Zorbax SB-C18 column (5 μm, 250 × 9.4 mm, 3 ml/min) and an analytical Zorbax SB-C18 column (5 μm, 250 × 4.6 mm, 1 ml/min), respectively. TLC was run for monitoring collected fractions on silica gel GF254 plates (Qingdao Marine Chemical Co., Ltd., China). The visualization of spots on the plates were conducted by using an ultraviolet lamp at the wavelength of 254 nm or by heating with H2SO4-EtOH (5%).
3.2 Plant material
The M. chinensis twigs were harvested in August 2019 from Honghe Hani and Yi Autonomous Prefecture (Yunnan, China), with the authentication of Dr. Jindong Zhong (Kunming University of Science and Technology). A voucher specimen (serial number: KMUST201903) was stored at the Department of Life Science and Technology.
3.3 Extraction and purification
The air-dried powdered of M. chinensis twigs (15 kg) was extracted with 70% acetone/H2O by refluxing for 24 h (30 L × 3 times). After filtration and evaporation procedures, the extract (1286 g) was yielded and thoroughly dissolved in H2O. The mixture was then extracted by petroleum ether, chloroform, ethyl acetate, and n-butanol, respectively. The ethyl acetate extract (145 g) was separated to four fractions (Fr. A–D) by silica gel column (20 × 300 cm) eluted with dichloromethane-methanol (1:0–0:1).
Fr. B (32.0 g) was subjected to four fractions (Fr. B-1–B-4) through MCI (90% MeOH/H2O) and RP-18 eluting with MeOH/H2O (30–100%). Fr. B-2 (1.5 g) was separated by silica gel column eluted with chloromethane-methanol (40:1–2:1) to give 9 (5.4 mg) and 23 (33.5 mg). Fr. B-3 (10.0 g) was separated to four fractions (Fr. B-3-1–B-3-5) by ODS C-18 (MeOH/H2O, gradient 20%–100%). Fr. B-3-2 (1.3 g) was subjected to silica gel column eluted with dichloromethane-methanol (15:0–1:1) to obtain compounds 2 (3.2 mg) and 7 (4.2 mg). Compounds 5 (20.3 mg), 17 (2.7 mg, tR = 21.2 min), 26 (4.4 mg, tR = 12.5 min), and 34 (4.1 mg, tR = 15.8 min) were obtained from Fr. B-3-3 (2.3 g) by Sephadex LH-20 (MeOH) and semi-preparative HPLC (85% MeOH-H2O, 3 ml/min). Fr. B-3-4 (1.1 g) was purified by Sephadex LH-20 (CH2Cl2–MeOH, 1:1) and semi-preparative HPLC (73% MeOH–H2O, 3 ml/min) to yield 24 (5.8 mg, tR = 14.8 min), 25 (5.7 mg, tR = 16.0 min), and 33 (5.3 mg, tR = 10.6 min). Separation of Fr. B-3-5 (2.0 g) with silica gel column to obtain 22 (10.0 mg). Fr B-4 (1.0 g) was chromatographed over an ODS C-18 column (MeOH/H2O, 30–100%) and semi-preparative HPLC (68% MeOH–H2O, 3 ml/min) to give 1 (4.0 mg, tR = 8.0 min), 8 (1.6 mg, tR = 12.5 min), 11 (3.2 mg, tR = 17.0 min), 12 (3.1 mg, tR = 19.8 min), 31 (7.9 mg, tR = 22.6 min), and 32 (10.8 mg, tR = 16.0 min).
Fr. C (33.0 g) was fractioned by RP-18 (MeOH/H2O, gradient 30–100%) to obtain subfractions Fr. C-1–C-4. Fr. C-3 (5.4 g) was applied ODS C-18 column chromatography eluted with MeOH/H2O (30%–100%) and silica column (CH2Cl2-MeOH, 8:1–1:1) to give 10 (2.2 mg), 13 (9.2 mg), 20 (37.1 mg), and 35 (3.1 mg). Fr. C-4 (9.4 g) was chromatographed successively over silica gel (dichloromethane-methanol 10:1–1:1), Sephadex LH-20 gel (MeOH) and semi-preparative HPLC (65% MeOH–H2O, 3 ml/min) to give compounds 19 (14.1 mg, tR = 18.1 min), 21 (110.0 mg, tR = 16.5 min), and 14 (3.4 mg, tR = 13.2 min).
Fr. D (29.0 g) was fractioned via Sephadex LH-20 gel, eluting with MeOH to afford subfractions Fr. D-1–D-4. Fr. D-2 (1.2 g) was purified by silica gel column eluted with PE/EtOAc (15:1–1:1) and Sephadex LH-20 (MeOH), successively, yield compounds 3 (5.3 mg), 4 (6.2 mg), and 6 (7.3 mg). Compounds 15 (4.1 mg, tR = 14.6 min), 16 (9.3 mg, tR = 17.5 min), and 27 (7.2 mg, tR = 23.8 min) were obtained from Fr. D-3 (4.2 g) by Sephadex LH-20 (MeOH) and semi-preparative HPLC (52% MeOH–H2O, 3 ml/min). Fr. D-4 (11.2 g) was separated by silica gel column eluting with petroleum ether-ethyl acetate (10:1–1:1) to give Fr. D-4-1–D-4-3. Compound 28 (7.1 mg, tR = 13.5 min) was obtained from Fr. D-4-2 (334.6 mg) by semi-preparative HPLC (85% MeOH–H2O, 3 ml/min). Fr. D-4-3 (2.7 g) was applied to ODS column chromatography eluted with MeOH/H2O (30–100%) and silica column eluting with chloromethane-methanol (30:1–1:1) to give 18 (12.1 mg), 29 (8.2 mg), and 30 (4.2 mg).
3.3.1 Acacetin 7-O-[β-D-apiofuransyl-(1ʹʹʹ → 4ʹʹ)]-β-D-xylopyranoside (1)
Amorphous powder; [α]25 D = − 25.1 (c = 0.10, MeOH); IR (KBr) vmax 3495, 3374, 2917, 2866, 1660, 1604, 1583, 1497, 1428, 1377, 1239, 1025, and 834 cm−1; UV (MeOH): λmax (log ε) 324 (2.23) nm; HRESIMS m/z 571.1408 [M + Na] + (calcd for C26H28O13Na, 571.1428); 1H and 13C NMR data (Table 1).
3.3.2 Acacetin 7-O-[4ʹʹʹ-O-acetyl-β-D-apiofuransyl-(1ʹʹʹ → 2ʹʹ)]-6ʹʹ-O-acetyl-β-D-glucoside (2)
Amorphous powder; [α]25 D = − 28.0 (c = 0.10, DMSO); IR (KBr) vmax 3430, 2921, 2850, 1728, 1615, 1587, 1489, 1365, 1300, 1182, 1080, 987, and 829 cm−1; UV (MeOH): λmax (log ε) 327 (3.23) nm; HRESIMS m/z 663.1910 [M + H] + (calcd for C31H34O16, 663.1920); 1H and 13C NMR data (Table 1).
3.3.3 3ʹ, 4ʹ-Dimethoxyluteolin 7-O-[β-D-apiofuransyl-(1ʹʹʹ → 4ʹʹ)]-β-D-xylopyranoside (3)
Amorphous powder; [α]25 D = − 35.0 (c = 0.10, DMSO); IR (KBr) vmax 3389, 2928, 2865, 1700, 1659, 1608, 1514, 1382, 1338, 1298, 1183, 1123, 1085, 987, and 830 cm−1; UV (MeOH): λmax (log ε) 326 (2.37) nm; HRESIMS m/z 579.1717 [M + H] + (calcd for C27H30O14, 579.1714); 1H and 13C NMR data (Table 1).
3.3.4 9ʹ,9ʹʹ-dimethyl clinopodic acid C (19)
White amorphous powder; [α]25 D = + 100.0 (c = 0.10, MeOH); IR (KBr) vmax 3431, 3040, 2954, 2925, 2851, 1741, 1607, 1585, 1506, 1440, 1269, 1117, 978, 858, and 812 cm−1; UV (MeOH): λmax (log ε) 327 (3.14) nm; HRESIMS m/z 589.1293 [M + Na] + (calcd for C29H26O12Na, 589.1322); 1H and 13C NMR data (Table 2).
3.3.5 Mosla chinensis glycoside B1 (27)
White amorphous powder; [α]25 D = -37.8 (c = 0.10, MeOH); IR vmax 3501, 3407, 3305, 2873, 1610, 1518, 1365, 1035, 826, and 714 cm−1; ECD (MeOH) λmax (Δε) 211 (-4.82), 247 (+ 2.43) nm; UV (MeOH): λmax (log ε) 292 (2.57) nm; HRESIMS, m/z 447.1638 [M + Na] + (calcd for C21H28O9Na, 447.1631); 1H and 13C NMR data (as shown in Table 2).
3.3.6 2,5-dimethoxyphenethyl 3,4,5-trimethoxybenzoate (28)
Colorless crystal; [α]25 D = − 12.3 (c = 0.10, MeOH); IR vmax 3445, 2933, 2843, 1715, 1592, 1512, 1460, 1415, 1334, 1228, 1127, 1028, 1002, 862, 807, and 764 cm−1; UV (MeOH) λmax (log ε) 310 (2.78) nm; HRESIMS, m/z 377.1592 [M + H] + (calcd for C20H24O7, 377.1595); 1H and 13C NMR data (as shown in Table 2).
3.3.7 4-(hydroxy(2-(3-hydroxy-4-(hydroxymethyl) phenyl) propoxy) methyl)-2-methy-lphenol (29)
White amorphous powder; [α]25 D = − 12.4 (c = 0.10, MeOH); IR vmax 3415, 2921, 2853, 1512, 1456, 1407, 1263, 1170, 1063, and 1023 cm−1; UV (MeOH): λmax (log ε) 251 (2.25) nm; HRESIMS, m/z 319.1537 [M + H] + (calcd for C18H22O5, 319.1540); 1H and 13C NMR data (as shown in Table 2).
3.4 Acid hydrolysis and determination of the absolute configuration of sugars
Based on the method reported by Wu et al. [47], the D glucopyranose configurations in compounds 1–3 and 27 were measured. Compounds 1–3 and 27 (1 mg per compound) were individually mixed with 3 ml HCl (2 M). Each mixture was boiled for 4 h at 100 ℃. After neutralization with NaHCO3, the mixture was treated by EtOAc. Subsequently, the H2O layer was evaporated and dissolved in DMSO (1.0 ml) before the acetic anhydride (40 μL) and 1-Methylimidazole (20 μL). After the reaction, extracted with EtOAc and analysed by GC. Monosaccharide compositions in compounds were identified by coeluting with authentic monosaccharide.
3.5 ECD quantification
The conformational structures of compounds were achieved from Chem3D modeling and ROESY spectra. In terms of the conformations, low-energy conformers of 27 were created via CONFLEX software by using an energy window (10 kcal/mol, MMFF94S) [48]. Density functional theory (DFT) method was employed to optimize the selected conformers in MeOH at the B3LYP/6-31 G (d) level [49]. Geometry optimizations and predictions of the conformersʹ ECD spectra were conducted by TD-DFT-B3LYP/6-311G (2d, p) level using a solvent (IEFPCM solvent model for methanol) [50]. SpecDis 1.71 was hired to generate the predicted curves of ECD, and Gaussian 16 package was applied to all predictions [51]. After UV correction, compound 27 spectrum was weighted using the Boltzmann distribution.
3.6 Anti-influenza virus assay
Based on the approach described by Dang et al. [52], anti-influenza virus assay was carried out. Briefly, prior to infection, MDCK cells (8 × 103 cells/well) were cultivated for 24 h in 96-well plates, and the medium was removed. The mixture of compounds (at 3.125, 6.25, 12.5, 25, and 50 μM) and H1N1 virus was cultured at ambient temperature for 15 min and then transferred to the plates containing MDCK cells. The plates were stored at 37 ℃ with 5% CO2 for 48 h. Subsequently, the antiviral activity was quantified using microscopy. The obtained antiviral activity was verified using the CellTiter-Glo luminescent cell viability assay (Promega, #G7570). Each compound's cytotoxicity was evaluated through incubating with uninfected MDCK cells for 48 h [53, 54].
3.7 Western blot assay
Influenza virus-infected MDCK cells were added with compound 20 (50 μM) at distinct time points (2, 5, 8, and 10 h). Cell lysates were harvested, and proteins from the supernatant were obtained [55]. After being electrophorized on a 12% SDS-PAGE electrophorized gel, protein extracts were distributed on a PVDF membrane and then cultivated for 1 h in blocking media (5% nonfat milk) at ambient temperature. Immunoblotting was performed using antibodies: anti-β-actin (SC-47778 (Catalog No.), Santa Cruz) and anti-nucleoprotein (EPR25683 (Abcam No.), GeneTex). The target proteins were visualized by chemiluminescence (ECL, Beyotime).
3.8 Immunofluorescence assay
MDCK cells (1 × 105 cells) were loaded into each well of 24-well plates. The plates were kept under 5% CO2 condition at 37 ℃. As the cells increased by 50%, the cells were added and cultured with A/WSN/33/2009 (H1N1) virus (MOI = 0.1) for 2 h. After the removal of the supernatant, the cells were rinsed two times using PBS. Subsequently, the 20 was supplemented to cells and stored under conditions of 5% CO2 and 37 ℃. At 2, 5, 8, and 10 h of incubation, the cells were fixed using PFA in PBS (4%, Beyotime Biotechnology) at refrigerated temperature for 10 min. The fixed cells were then permeabilized for 10 min at room temperature using 0.1% Triton X-100 in PBS and blocked for 1 h at 37 ℃ using 3% BSA in PBS. Afterward, the treated cells were first stored at 4℃ overnight supplemented with nucleoprotein antibody diluted in 3% BSA (1:250, Abcam, CA, USA) and then cultured at ambient temperature for 1 h with fluorescein isothiocyanate (FITC)-labeled secondary antibody diluted in 3% BSA (1:250). The nucleus in cells was stained by DAPI for 10 min at ambient temperature. After staining, the fluorescence was examined by an inverted fluorescence microscope (Nikon A1R/A1, Shanghai, Japan) [56].
3.9 Hemagglutination inhibition assay
Hemagglutination inhibition (HAI) test was utilized to evaluate the activity of 20 against HA-mediated avian RBCs hemagglutination [57]. Briefly, 20 (10, 20, 40, and 80 μM) with influenza A/WSN/33/2009 (H1N1) (2–6 hemagglutination titer) was supplemented into 96-well plates and cultured under an ambient condition for 1 h. Afterward, 1% chicken RBCs saline solution (50 μL) was loaded into each well. After incubation for 30 min at ambient temperature, the hemagglutination was examined.
3.10 Molecular docking
The protein structure of H1N1 HA (PDB ID: 6CFG) was achieved from the RCSB protein data bank. Chemdraw3D was hired to construct 20's 3D structures. The docking procedures were provided by the AutoDock Software with a graphics interface (AutoGrid/AutoDock 4.2.6 and Vina). With the AutoDock tools, the deletion of all water molecules was executed, while the refined model was added with the polar hydrogen atoms and charges. Then, docking was conducted using AutoDock/Vina based on the HA information and the grid box characteristics of the studied compound in the configuration file. In the process of docking, the 20 structure and HA protein structure were regarded as rigid [58, 59].
3.11 Nitric oxide production assay
Based on the previously reported approach [60, 61], NO production in cells was assayed. Cells were loaded into 96-well plates (8×104/ well). The cells were treated by compounds (at 3.125, 6.25, 12.5, 25, and 50 μM) for 1 h, supplemented with LPS (1 μg/ml), and cultivated for 24 h. After treatment, a microplate reader (Thermo Fisher Scientific, Massachusetts, USA) was hired to quantify the absorbance values with the wavelength of 540 nm. The positive and negative controls were separately L-NMMA and DMSO.
4 Conclusions
This work systematically explored the phytochemical characteristics of thirty-five compounds extracted from M. chinensis twigs. The compounds included three unreported flavonoids (1–3), one undescribed phenylpropanoid (19) and three new monoaromatic hydrocarbons (27–29), and 28 known compounds. Compound 27's absolute configuration was interpreted and visualized with the assistance of ECD calculation. Compound 20 exhibited the most significant activity against A/WSN/33/2009 (H1N1) virus (IC50 = 20.47 μM). Further research showed that 20 could bind to influenza virus surface antigen HA1 and inhibit the early adsorption process of the influenza A/WSN/33/2009 (H1N1) virus strain. Furthermore, compounds 9, 22, 23, and 25 displayed moderate inhibitory effects on the NO expression in LPS inducing Raw 264.7 cells with IC50 values of 22.78, 20.47, 27.66, and 30.14 µM, respectively.
The effect of M. chinensis on influenza virus infection, controlling the adsorption of virus and excessive inflammatory reaction in the infection process. Our findings will enrich the study of the structural diversity of M. chinensis and provide insights into understanding the plant's anti-influenza function, which may launch scientific basis for the following research about the development of antiviral beverages and resources utilization of M. chinensis.
Notes
Acknowledgements
This paper was financed by National Natural Science Foundation of China (No. 31660100), Innovative Team of Yunnan Province (No. 2019HC018), the key Project of Yunnan Province (No. 202103AC10005, No. 202302AG050004), and Higher Educational Key Laboratory for New drugs for Viral Respiratory Diseases (Chinese Traditional Medicine) of Yunnan Province.
Author contributions
S.-Y. F. isolated and identified of the compounds; writing—original draft. N. J. was responsible for the biological activities assessment. J.-Y. Y. performed chemical calculation and wrote the paper. L.-Y. Y. and J.-C. D. contributed to the extraction, isolation, and identification of the compounds. D.L. and J.-D. Z. designed experiments. X.-Q. C. and R.-T. L. checked the whole manuscript. All authors read and approved the fnal manuscript.
Data availability
The datasets used or analysed during the current study are available from the corresponding author on reasonable request.
Declarations
Competing interests
The authors declare that there are no competing interests associated with this work.
References
-
1.Heo JY, Song JY, Noh JY, Choi MJ, Yoon JG, Lee SN, Cheong HJ, Kim WJ. Effects of influenza immunization on pneumonia in the elderly. Hum Vacc Immunother 2018;14: 744-9. CrossRef PubMed Google Scholar
-
2.Schanzer DL, Langley JM, Tam TWS. Hospitalization attributable to influenza and other viral respiratory illnesses in Canadian children. Pediatr Infect Dis J 2006;25: 795-800. CrossRef PubMed Google Scholar
-
3.Li Z, Wang H, Wang FX, Li HY, Cao F, Luo DQ, Zhang Q, Chen FL. Isolation of essential oil from Mosla chinensis Maxim by surfactant-enzyme pretreatment in high-solid system and evaluation of its biological activity. Ind Crop Prod 2022;189: 115871. CrossRef PubMed Google Scholar
-
4.National Pharmacopoeia Committee. Pharmacopoeia of People's Republic of China. Part 1. Medical Science and Technology Press: Beijing, 2015; 259–60. PubMed Google Scholar
-
5.Cao L, Si JY, Liu Y, Sun H, Jin W, Li Z, Zhao XH, Pan RL. Essential oil composition, antimicrobial and antioxidant properties of Mosla chinensis Maxim. Food Chem 2009;115: 801-5. CrossRef PubMed Google Scholar
-
6.Liu MT, Luo FY, Zeng JG. Composition analysis of essential oil of Mosla chinensis Maxim and its antibacterial and antioxidant activity. Chin Tradit Patent Med 2020;42: 3091-5. PubMed Google Scholar
-
7.Lin CL, Cai JZ, Lin GY. Chemical constituent study of volatile oils from the Mosla chinensis Maxim in Zhejiang Province. Chin Arch Tradit Chin Med 2012;30: 197-8. PubMed Google Scholar
-
8.Feng Y, Liu J. Effects of volatile oil from Mosla chinensis Maxim on bacteriostasis and immune response. Amino Acids Biotic Resour 2009;31: 30-2. PubMed Google Scholar
-
9.Ge B, Lu XY, Jiang HM. Study on antibacterial effect of volatile oil of Mosla chinensis Maxim in vitro. Chin J Tradit Veterinary Sci 2005;2: 8-10. PubMed Google Scholar
-
10.Zhang XX, Wu QF, Yan YL, Zhang FL. Inhibitory effects and related molecular mechanisms of total flavonoids in Mosla chinensis Maxim against H1N1 influenza virus. Inflamm Res 2018;67: 179-89. CrossRef PubMed Google Scholar
-
11.Zhang L, Yang LY, Li RT, Yu F, Zhong JD. A new prenylated 3-benzoxepin derivative with anti-influenza A virus activity from Elsholtzia penduliflora. Nat Prod Res 2022;36: 719-25. CrossRef PubMed Google Scholar
-
12.Yang LY, Du JC, Li RT, Yu F, Zhong JD. Bodiniosides S-Y, seven new triterpenoid saponins from Elsholtzia bodinieri and their anti-Influenza activities. Molecules 2021;26: 6535. CrossRef PubMed Google Scholar
-
13.Qiao Y, Sun WW, Wang JF, Zhang JD. Flavonoids from Podocarpus macrophyllus and their cardioprotective activities. J Asian Nat Prod Res 2014;16: 222-9. CrossRef PubMed Google Scholar
-
14.Sugimoto S, Yamano Y, Desoukey SY, Katakawa K, Matsunami K. Isolation of sesquiterpene–amino acid conjugates, onopornoids A-D, and a flavonoid glucoside from Onopordum alexandrinum. J Nat Prod 2019;82: 1471-7. CrossRef PubMed Google Scholar
-
15.Sinha NK, Seth KK, Pandey VB, Dasgupta B, Shah AH. Flavonoids from the flowers of Clerodendron infortunatum. Planta Med 1981;42: 296-8. CrossRef PubMed Google Scholar
-
16.Seo YH, Trinh TA, Ryu SM, Kim HS, Lee J. Chemical constituents from the aerial parts of Elsholtzia ciliata and their protective activities on glutamate-induced HT22 Cell Death. J Nat Prod 2020;83(10): 3149-55. CrossRef PubMed Google Scholar
-
17.Perry NB, Foster LM. Antiviral and antifungal flavonoids, plus a triterpene, from Hebe cupressoides. Planta Med 1994;60(5): 491-2. CrossRef PubMed Google Scholar
-
18.Hori K, Satake T, Saiki Y, Tanaka N, Murakami T, Chen CM. Chemical and chemotaxonomical studies of Filices. LXXIV. The novel flavanone glycosides of Pyrrosia linearfolia (HOOK.) Ching. J Pharm Soc Jpn 1988;108(5): 417-21. CrossRef PubMed Google Scholar
-
19.Zhang XL, Guo YS, Wang CH, Li GQ, Xu JJ, Chung HY, Ye WC, Li YL, Wang GC. Phenolic compounds from Origanum vulgare and their antioxidant and antiviral activities. Food Chem 2014;152: 300-6. CrossRef PubMed Google Scholar
-
20.Lai GF, Zhu XD, Luo SD, Wang YF. Chemical constituents from Elsholtzia rugulos. Chin Tradit Herb Drugs 2008;5(39): 661-4. PubMed Google Scholar
-
21.Oyama KI, Kondo T. Total synthesis of apigenin 7,4 prime-di-O-β-glucopyranoside, a component of blue flower pigment of Salvia patens, and seven chiral analogues. Tetrahedeon 2004;60(9): 2025-34. CrossRef PubMed Google Scholar
-
22.Besson E, Chopin J. Sugar ring isomerization in C-arabinosyl flavones. Phytochemistry 1983;22(9): 2051-6. CrossRef PubMed Google Scholar
-
23.Wang XF, Li H, Jiang K, Wang QQ, Zheng YH, Wei T, Tan CH. Anti-inflammatory constituents from Perilla frutescens on lipopolysaccharide-stimulated RAW 264.7 cells. Fitoterapia 2018;130: 61-5. CrossRef PubMed Google Scholar
-
24.Formisano C, Rigano D, Senatore F, Bancheva S, Maggio A, Rosselli S, Bruno M. Flavonoids in subtribe centaureinae (Cass.) Dumort. (Tribe Cardueae, Asteraceae): distribution and 13C-NMR spectral data. Chem Biodiversity 2012;9(10): 2096-158. CrossRef PubMed Google Scholar
-
25.Kong CH, Xu XH, Hu F, Chen XH, Ling B, Tan ZW. Using specific secondary metabolites as markers to evaluate allelopathic potentials of rice varieties and individual plants. Chin Sci Bull 2002;47(10): 839-43. CrossRef PubMed Google Scholar
-
26.Zhang Q, Guilhon CC, Fernandes PD, Boylan F. Antinociceptive and anti-inflammatory activities of Elsholtzia ciliata (Thunb.) Hyl. (Lamiaceae) extracts. Planta Med 2014;80(16): 1406-1406. CrossRef PubMed Google Scholar
-
27.Chen XF, Ma GX, Huang Z, Wu TY, Xu XD, Zhong XM. Identification of water-soluble phenolic acids from Clerodendranthus spicatus. Chin Tradit Herb Drugs 2017;48(13): 2614-8. PubMed Google Scholar
-
28.Tsai SF, Lee SS. Neolignans as xanthine oxidase inhibitors from Hyptis rhomboides. Phytochemistry 2014;101: 121-7. CrossRef PubMed Google Scholar
-
29.Lee C, Kim J, Lee H, Lee S, Kho Y. Two new constituents from Isodon excisus and their evaluation in an apoptosis inhibitioni assay. J Nat Prod 2001;64(5): 659-60. CrossRef PubMed Google Scholar
-
30.Su D, Tang W, Hu Y, Liu Y, Yu S, Ma S, Qu J, Yu D. Lignan glycosides from Neoalsomitra integrifoliola. J Nat Prod 2008;71(5): 784-8. CrossRef PubMed Google Scholar
-
31.Gu QC, Yin ZK, Feng ZM, Jiang JS, Zhang X, Zhang PC, Yang YN. Three 11,12-seco-tanshinone derivatives from the rhizomes of Salvia miltiorrhiza. J Asian Nat Prod Res 2020;22(10): 935-40. CrossRef PubMed Google Scholar
-
32.Kato H, Li W, Koike M, Wang Y, Koike K. Phenolic glycosides from Agrimonia pilosa. Phytochemistry 2010;71(16): 1925-9. CrossRef PubMed Google Scholar
-
33.Dhingra MS, Dhingra S, Kumria R, Chadha R, Singh T, Kumar A, Karan M. Effect of trimethylgallic acid esters against chronic stress-induced anxiety-like behavior and oxidative stress in mice. Pharmacol Rep 2014;66(4): 606-12. CrossRef PubMed Google Scholar
-
34.Takeda Y, Tomonari M, Arimoto S, Masuda T, Otsuka H, Matsunami K, Honda G, Ito M, Takaishi Y, Kiuchi F, Khodzhimatov OK, Ashurmetov OA. A new phenolic glucoside from an Uzbek medicinal plant, Origanum tyttanthum. J Nat Med 2008;62(1): 71-4. CrossRef PubMed Google Scholar
-
35.Bravo JA, Sauvain M, Gimenez A, Munoz V, Callapa J, Le L, Massiot G, Lavaud C. Bioactive phenolic glycosides from Amburana cearensis. Phytochemistry 1999;50(1): 71-4. CrossRef PubMed Google Scholar
-
36.Koike K, Li W, Liu LJ, Hata E, Nikaido T. New phenolic glycosides from the seeds of Cucurbita moschata. Chem Pharma Bull 2005;53(2): 225-8. CrossRef PubMed Google Scholar
-
37.Guetchueng ST, Nahar L, Ritchie KJ, Ismail FMD, Dempster NM, Nnanga EN, Sarker SD. Phenolic compounds from the leaves and stem bark of Pseudospondias microcarpa (A. Rich.) Engl. (Anacardiaceae). Biochem Syst Ecol. 2020;91: 104078. PubMed Google Scholar
-
38.Tagousop CN, Ngnokam D, Harakat D. Three new flavonoid glycosides from the aerial parts of Graptophyllum grandulosum Turril (Acanthaceae). Phytochem Lett 2017;19: 172-5. CrossRef PubMed Google Scholar
-
39.Xu JZ, Zhang SS, Qu HB. Chemical constituents from Viola yedoensis. Chin Tradit Herb Drugs 2010;41(9): 1423-5. PubMed Google Scholar
-
40.Miyazawa M, Hisama M. Antimutagenic activity of flavonoids from Chrysanthemum morifolium. Biosci Biotechnol Biochem 2003;67(10): 2091-9. CrossRef PubMed Google Scholar
-
41.Murata T, Sasaki K, Sato K, Yoshizaki F, Yamada H, Mutoh H, Umehara K, Miyase T, Warashina T, Aoshima H. Matrix metalloproteinase-2 inhibitors from Clinopodium chinense var. parviflorum. J Nat Prod 2009;72(8): 1379-84. CrossRef PubMed Google Scholar
-
42.Zhong M, Sun G, Zhang X, Sun G, Xu X, Yu S. A New prenylated naphthoquinoid from the aerial parts of Clinopodium chinense (Benth.) O. Kuntze. Molecules 2012;17(12): 13910-6. CrossRef PubMed Google Scholar
-
43.Hiipakka RA, Zhang HZ, Dai W, Dai Q, Liao S. Structure-activity relationships for inhibition of human 5alpha-reductases by polyphenols. Biochem Pharmacol 2002;63(6): 1165-76. CrossRef PubMed Google Scholar
-
44.Liu F, Zhong J, Zhou Y, Gao Z, Walsh PJ, Wang X, Ma S, Hou S, Liu S, Wang M, Wang M, Bian Q. Cobalt-catalyzed enantioselective negishi cross-coupling of racemic α-Bromo esters with arylzincs. Chemistry 2018;24(9): 2059-64. CrossRef PubMed Google Scholar
-
45.Mair CM, Ludwig K, Herrmann A, Sieben C. Receptor binding and pH stability—how influenza A virus hemagglutinin affects host-specific virus infection. Bba-Biomembranes 2014;1838(4): 1153-68. CrossRef PubMed Google Scholar
-
46.Tavares LP, Teixeira MM, Garcia CC. The inflammatory response triggered by influenza virus: a two edged sword. Inflamm Res 2017;66(4): 283-302. CrossRef PubMed Google Scholar
-
47.Wu ST, Li F, Wang YX, Yu BH, Ma CL, Qiu HQ, Wang GS. Phenylpropanoids from Brachybotrys paridiformis Maxim. Ex Oliv. and their anti-HBV activities. Phytochemistry 2022;197: 113114. CrossRef PubMed Google Scholar
-
48.Fujihara T, Obora Y, Tokunaga M, Sato H, Tsuji Y. Dendrimer N-heterocyclic carbene complexes with rhodium(Ⅰ) at the core. Chem Commun 2005;8(36): 4526-8. CrossRef PubMed Google Scholar
-
49.Li JC, Dai WF, Liu D, Jiang MY, Zhang ZJ, Chen XQ, Chen CH, Li RT, Li HM. Bioactive ent-isopimarane diterpenoids from Euphorbia neriifolia. Phytochemistry 2020;175: 112373. CrossRef PubMed Google Scholar
-
50.Chen X, Cao YG, Ren YJ, Liu YL, Fan XL, He C, Li XD, Ma XY, Zheng XK, Feng WS. Ionones and lignans from the fresh roots of Rehmannia glutinosa. Phytochemistry 2022;203: 113423. CrossRef PubMed Google Scholar
-
51.Wang P, Liu F, Yang X, Liang Y, Li S, Su G, Jin DQ, Ohizumi Y, Xu J, Guo Y. Clerodane diterpenoids from Scutellaria formosana with inhibitory effects on NO production and interactions with iNOS protein. Phytochemistry 2017;144: 141-50. CrossRef PubMed Google Scholar
-
52.Dang Z, Jung K, Zhu L, Lai W, Xie H, Lee KH, Huang L, Chen CH. Identification and synthesis of quinolizidines with anti-influenza a virus activity. ACS Med Chem Lett 2014;5(8): 942-6. CrossRef PubMed Google Scholar
-
53.Vanderlinden E, Göktas F, Cesur Z, Froeyen M, Reed ML, Russell CJ, Cesur N, Naesens L. Novel inhibitors of influenza virus fusion: structure-activity relationship and interaction with the viral hemagglutinin. J Virol 2010;84(9): 4277-88. CrossRef PubMed Google Scholar
-
54.Jones JC, Turpin EA, Bultmann H, Brandt CR. Inhibition of influenza virus infection by a novel antiviral peptide that targets viral attachment to cells. J Virol 2006;80(24): 11960-7. CrossRef PubMed Google Scholar
-
55.Zhang YZ, Naguro I, Herr AE. In situ single-cell western blot on adherent cell culture. Angew Chem Int Ed Engl 2019;58(39): 13929-34. CrossRef PubMed Google Scholar
-
56.Liang XX, Zhang XJ, Zhao YX, Feng J, Zeng JC, Shi QQ, Kaunda JS, Li XL, Wang WG, Xiao WL. Aspulvins A-H, aspulvinone analogues with SARS-CoV-2 M(pro) inhibitory and anti-inflammatory activities from an Endophytic Cladosporium sp. J Nat Prod 2022;85(4): 878-87. CrossRef PubMed Google Scholar
-
57.Zhang T, Lo CY, Xiao M, Cheng L, Pun Mok CK, Shaw PC. Anti-influenza virus phytochemicals from Radix Paeoniae Alba and characterization of their neuraminidase inhibitory activities. J Ethnopharmacol 2020;253: 112671. CrossRef PubMed Google Scholar
-
58.Shi WZ, Jiang LZ, Song GP, Wang S, Xiong P, Ke CW. Study on the antiviral activities and hemagglutinin-based molecular mechanism of novel chlorogenin 3-O-β-chacotrioside derivatives against H5N1 subtype viruses. Viruses 2020;12(3): 304. CrossRef PubMed Google Scholar
-
59.Ye M, Liao Y, Wu L, Qi W, Choudhry N, Liu Y, Chen W, Song G, Chen J. An oleanolic acid derivative inhibits hemagglutinin-mediated entry of influenza A virus. Viruses 2020;12(2): 225. CrossRef PubMed Google Scholar
-
60.Lee JW, Jin Q, Jang H, Lee D, Han SB, Kim Y, Hong JT, Lee MK, Hwang BY. Jatrophane and ingenane-type diterpenoids from Euphorbia kansui inhibit the LPS-induced NO production in RAW 264.7 cells. Bioorg Med Chem Lett 2016;26(14): 3351-4. CrossRef PubMed Google Scholar
-
61.Cao L, Li RT, Chen XQ, Xue Y, Liu D. Neougonin A inhibits lipopolysaccharide-induced inflammatory responses via downregulation of the NF-kB signaling pathway in RAW 264.7 macrophages. Inflammation 2016;39: 1939-48. CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2024
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
