


α-Glucosidase inhibitive diarylheptanoids from Ottelia acuminata var. acuminata, a traditional vegetable of Bai Nationality in Yunnan

Abstract
Diabetes is an urgent health issue characterized by ethnic and regional variations, and is inseparable from the different dietary habits. It is worthy to note that the incidence of diabetes in Bai nationality has been reported to be much lower than Han in China. As a daily vegetable of Bai, the phytochemical and antidiabetic study of Ottelia acuminata var. acuminata had not been carried out. In this study, 41 metabolites with diverse diarylheptanoid (six new ones, Otteacumienes A-F), flavone, sesquiterpenoid, coumarin, lignan, polyacetylene, and alkaloid skeletons were characterized from O. acuminata var. acuminata. Among them, the racemic nature of 3 was characterized by chiral resolution and calculated ECD methods. The biological study revealed diarylheptanoids showed significant α-glucosidase inhibitory activities with 5 as the most effective one (60-fold stronger than acarbose). Molecular docking studies indicated that these structures have different binding cavities with acarbose. This study demonstrated that O. acuminata var. acuminata might correlated with the low incidence diabetes of Bai and the diarylheptanoids may have potential therapeutic value for diabetes mellitus.Graphical Abstract

Keywords
Ottelia acuminata var. acuminata Bai nationality Vegetable Diarylheptanoids α-glucosidase1 Introduction
The latest research from IDF (International Diabetes Federation) revealed that about 8.8% of the world's population suffers from diabetes disease. Among them, type 2 diabetes (T2DM) is the most common type, accounting for more than 90% of all diabetic cases worldwide [1]. Treatment of T2DM requires both diet and exercise to address the overweight or obesity. A range of combination therapy options were available for T2DM such as sulfonylureas, α-glucosidase inhibitors (AGI), thiazolidinediones, dipeptidyl peptidase 4 (DPP-4) inhibitors, glucagon-like peptide 1 (GLP-1) agonists, and sodium-glucose co-transporter 2 (SGLT-2) inhibitors [2]. Among them, α-glucosidase is one of the most important digestive enzymes in the human body involved in the final step of carbohydrate digestion, and its inhibitors contributes to control postprandial glucose for diabetes treatment [3]. However, the common side effects of flatulence, diarrhea, and hepatotoxicity for the clinical α-glucosidase inhibitors (including acarbose and miglitol) encouraged us to find new type of inhibitors with high safety and efficiency [4].
It's well-know that the development of diabetes is closely relevant to the dietary and lifestyle. Interestingly, the investigation of the incidence of diabetes in China has showed the characteristics of ethnic and regional variations [5], especially for the Bai nationality (7.83%) in contrast to the Han nationality (11.83%) in rural Yunnan [6]. As one of the major ethnic minorities in Yunnan, the Bai nationality has a long history and splendid culture, most of whom live in Dali Bai Autonomous Prefecture [7]. Notably, O. acuminata var. acuminata had long been used as a traditional daily vegetable in Bai as record in the "Textual Research on Reality and Titles of Plants" published in 175 years ago [8].
As an edible unique plant in China, Ottelia acuminata var. acuminata belongs to the genus Ottelia of the Hydrocharitaceae family, and mainly distributed in Yunnan Province [9]. This plant is also a famous aquatic ornamental plant, attracting millions of tourists to Erhai and Lugu Lakes every year during the blooming period from May to October. Additionally, the high requirements for water quality could be applied to monitor water pollution and environmental protection [10]. In order to explore antidiabetic chemical constituents of a daily vegetable for Bai nationality, O. acuminata var. acuminata was selected for this study. However, except for a preliminary analytical investigation by HPLC-ESI-MS, the phytochemical and antidiabetic ingredients of this vegetable have not been reported so far [11].
In this study, the first study of phytochemistry on O. acuminata var. acuminata was conducted and 41 compounds including six new diarylheptanoids (Otteacumienes A-F, 1-6) and 35 known ones possessing diverse diarylheptanoid, flavone, sesquiterpenoid, coumarin, lignan, and polyacetylene skeletons (7-41) were obtained (Fig. 1). Structurally, the six new diarylheptanoids could be classified into three different types: diarylether, biaryl, and linear types of diaryheptanoids. Being distinct from diarylheptanoids reported from families Officinarum, Katsumadai, Blepharocalyx, Zingiberaceae, and Betulaceae [12], these structures in this study are characterized by low degree of oxidation. Bioactive study of compounds 3-8 showed substantial inhibitory effects on α-glucosidase but negligible effects on PTP1B, suggesting that they might be selective inhibitors of α-glucosidase. In addition, the molecular docking studies implied that different types diarylheptanoids are binding to the different sites of α-glucosidase, and the phenolic hydroxyl groups on diaryheptanoids might play a key role for their inhibitory activity.

Structures of new diarylheptanoids (1-6) and other representative compounds
2 Result and discussion
The DCM fraction of the EtOAc extract of O. acuminata var. acuminata was subjected to MCI-gel column, silica gel column chromatography, RP-C18 column chromatography, and preparative HPLC to afford eight diarylheptanoids including six new diarylheptanoids (Otteacumienes A-F, 1-6) and 35 known compounds possessing different phenylpropionids, coumarins, lignins, flavonoids, polyacetylene, sesquiterpenoid, and alkaloid architectures. Their structures were elucidated by comprehensive methods including NMR, MS, X-ray diffraction analyses, and calculated ECD spectra. In the antidiabetic studies, the diarylheptanoids showed significant α-glucosidase inhibitory activities and negligible effects on PTP1B. Additionally, molecular docking study further illustrated the possible binding cavities of the diarylheptanoids.
2.1 Structural identification of compounds
Otteacumiene A (1) was isolated as a yellow crystal and its molecular formula was determined as C19H18O3 by the HRESIMS at m/z 293.1182 [M-H]- (calculated for 293.1178). The IR spectrum showed characteristic hydroxyl broad absorption band at 3442 cm-1 and aromatic ring absorption band at 1632, 1590, 1519, 1428 cm-1. The 1H-NMR signals at δH 5.36 (1H, d, J = 2.2 Hz, H-3), 6.57 (1H, dd, J = 8.0, 2.2 Hz, H-5), 6.68 (1H, d, J = 8.0 Hz, H-6), δH 7.23 (1H, dd, J = 8.2, 2.6 Hz, H-2′), 7.29 (1H, dd, J = 8.2, 2.3 Hz, H-3′), 7.34 (1H, dd, J = 8.3, 2.3 Hz, H-5′), and 7.05 (1H, dd, J = 8.3, 2.6 Hz, H-6′) suggested a 1, 2, 4-trisubstituted benzene of an ABX spin system and a 1′, 4′-disubstituted aromatic rings of an AA′BB′ spin system (Table 1), respectively [13]. In addition, a cis and a trans carbon-carbon double bonds were evidenced by key 1H NMR data of δH 5.40 (1H, t, J = 10.1 Hz, H-9), 5.90 (1H, t, J = 10.1 Hz, H-10), 5.33 (1H, m, H-11), and 6.07 (1H, dt, J = 15.0, 4.0 Hz, H-12). Besides 16 carbon atoms of two benzene ring and two carbon-carbon double bonds, the remaining three carbon atoms were attributed to be two methylene (C-7 and C-13) and an oxygenated methine (C-8) according to the 13C-NMR and DEPT spectra (Table 2). The above characteristic signals implied that 1 could be a diaryheptanoid derivatives [14].
H NMR data of compounds 1-6a
13C NMR and DEPT (150 MHz) data of 1-6a
In the 2D NMR spectra, the key 1H-1H COSY correlations of H-7 (δH 2.34, d, J = 4.5 Hz)/H-8/H-9/H-10/H-11/H-12/H-13 (δH 3.51, m), together with the obvious HMBC correlations from H2-7 to C-3/C-5 and H2-13 to C-3′/C-5′/C-11 confirmed the presence of oxygenated unsaturated heptane chain and the connection of two benzene ring. The linkage of C-2 and C-1′ through an oxygen atom was elucidated by the downfield chemical shift of C-1 (δC 145.0), C-1′ (δC 157.6), and C-2 (δC 152.3), the key HMBC correlations from 1-OH to C-1/C-2/C-6, together with the degrees of unsaturation. (Fig. 2). Finally, the absolute configuration of C-8 was undoubtedly determined to be 8S by X-ray diffraction analysis using Cu Kα radiation. [Flack parameter = 0.08(4)] (Fig. 3, CCDC 2156830).

Key 1H-1H COSY and HMBC correlations of 1, 3, 4, and 5

X-ray crystallographic structures for 1 and 3
Otteacumienes B (2) was isolated as yellow oil. The molecular formula was determined to be the same as 1 by HREIMS. Detailed analysis of the 1H and 13C NMR spectra indicated that the structure of 2 was similar with that of 1 (Tables 1, 2). For 2, the main difference from 1 was ascribed to the position of carbon-carbon bond (Δ8, 9 and Δ10, 11) and hydroxyl (at C-12) in the heptane chain as deduced by the 1H-1H COSY correlations of H-7 (δH 2.96, m)/H-8 (δH 5.52, m)/H-9 (δH 5.69, dd, J = 15.6, 11.0 Hz)/H-10 (δH 5.96, t, J = 11.2 Hz)/H-11 (δH 5.23, t, J = 11.2 Hz)/H-12 (δH 4.46, td, J = 10.3, 3.2 Hz)/H-13 (δH 3.15, dd, J = 12.0, 3.2 Hz). The absolute configuration of C-12 was identified as 12S by comparison experimental with calculated ECD spectra (Fig. 4).

Calculated and experimental ECD spectra of 2 and 3
Otteacumiene C (3) was isolated as a colorless needle crystal. Its molecular formula was identified as C20H22O4 by the HRESIMS data at m/z 349.1407 [M + Na]+ (calcd for 349.1410). Detailed comparison of their 1D NMR data indicated 3 and 1 are structurally similar (Tables 1, 2). The most obvious differences of 3 compared to that of 1 lie in the appearance of an additional methoxy signal at δH 3.20/δC 57.4 as well as the absence of signals for a double bond in the heptane chain. These deductions were confirmed by downfield chemical shifts C-7 (δC 87.5) and C-8 (δC 75.0), the HMBC correlations from H-7 to C-3/C-4/C-5/C-9 and H-13 to C-3′/C-4′/C-5′/C-11, and COSY correlations of H-7/H-8/H-9/H-10/H-11/H-12/H-13 (Fig. 2). In addition, the crystals for single-crystal X-ray diffraction (Fig. 3, CCDC 2155136) were obtained, which clarified the relative configuration and the racemic nature of 3 with the crystal space group P2/n. After attempts with various chiral columns and conditions of mobile phase, the chiral separation of 3 was achieved on a chiral-phase HPLC apparatus using a DAICEL CORPORATION semi-preparative column (Fig. S2). To further determine the absolute configurations of enantiomers, quantum-chemical calculation method was used, which eventually assigned the absolute configurations of (+)-3 and (-)-3 to be 7S, 8S and 7R, 8R, respectively (Fig. 4).
Otteacumiene D (4) was also isolated as yellow oil. The molecular formula was established as C19H18O3 from its HRESIMS data at m/z 293.1182 [M-H]- (calculated 293.1187). The IR bands at 1508, 1612, 1703, 3390 cm-1 suggested the existence of aromatic ring, carbonyl, and hydroxyl groups in the structure. The 1H NMR spectrum showed two sets of 1, 2, 4-trisubstituted benzene rings signals at δH 7.28 (1H, d, J = 2.4 Hz, H-3′), 6.98 (1H, dd, J = 8.3, 2.4 Hz, H-5′), 6.72 (1H, d, J = 8.3 Hz, H-6′) and 6.87 (1H, d, J = 2.5 Hz, H-3), 7.03 (1H, dd, J = 8.4, 2.5 Hz, H-5), 6.85 (1H, d, J = 8.4 Hz, H-6), together with a pair of trans double bond signals at δH 5.54 and 5.79 (1H, dt, J = 14.5, 6.4 Hz, H-10 and 1H, dt, J = 14.5, 6.9 Hz, H-11) (Table 1). The 13C NMR and DEPT spectra displayed 19 carbon signals attributing to seven quaternary carbons (one carbonyl), eight methine, and four methylene. The evidences mentioned above, conjugated with a pair of unusual high field aromatic quaternary carbon signals at δC 128.3/ 126.6 (C-2/C-2′) suggest that 4 could be a biaryl type cyclic diaryheptanoid derivative (Tables 1, 2) [13, 14]. The 1H-1H COSY correlations of H-9 (δH 3.48, d, J = 6.4 Hz)/H-10/H-11/H-12 (δH 2.41, m)/H-13 (δH 2.76, J = 7.3, 5.8 Hz) and HMBC correlations from H2-7 to C-3/C-5/C-9, from H-10 to C-8/C-12, and from H2-13 to C-3′/C-4′/C-5′ established its biaryl type cyclic diaryheptanoid architecture. Then, the connection between C-2 and C-2′ was confirmed by the degrees of unsaturation together with the HMBC correlations of H-3/C-2′ and H-3′/C-2 (Fig. 2). Therefore, the structure of 4 was elucidated.
Otteacumiene E (5) was obtained as green oil, its molecular formula was determined to be C19H20O2, based on HRESIMS spectrum at m/z 279.1390 [M-H]- (calculated 279.1385). The 1H NMR spectrum revealed the presence of two sets of 1, 4-disubstituted benzene rings δH 7.00 (2H, m, H-3, 5), 6.75 (2H, m, H-2, 6), 7.04 (2H, m, H-3′, 5′), 6.74 (2H, m, H-2′, 6′), a pair of trans carbon-carbon double bond (δH 5.74, 1H, dt, J = 15.0, 7.0 Hz, H-8 and 6.38, 1H, m, H-9), and a pair of cis carbon-carbon double bond (δH 5.96 t, J = 11.0, Hz, H-10 and 5.35, 1H, dt, J = 11.0, 7.5 Hz, H-11) (Table 1) for a typical linear diaryheptanoid [13, 14]. The 13C NMR and DEPT spectroscopic data demonstrated 19 carbon signals attributing to four quaternary carbons, 12 methine, and three methylene (Table 2). In the 2D NMR spectra, the key 1H-1H COSY correlations of H-7/H-8/H-9/H-10/H-11/H-12H-13 and HMBC correlations from H2-7 to C-3/C-5 and H2-13 to C-3′/C-5′/C-11 confirmed diaryheptanoid nature of 5. In addition, the location of two hydroxyl at C-1 and C-1′ was evidenced by the HMBC correlations from 1-OH to C-2/C-6 and from 1′-OH to C-2′/C-6′ (Fig. 2). Therefore, the structure of 5 was established as shown.
Compound 6 was also isolated as green oil, showed a molecular ion at m/z 293.1547 [M-H]- in the HRESIMS (calculated 293.1542), which correlates to the molecular formula C20H22O2. The 1H and 13C NMR spectra were similar with those of 5 (Tables 1, 2) except for an extra methoxy signal (δH 3.75, δC 55.3) in 6. And this methoxyl was deduced to be located at C-1′ by its HMBC correlation with C-1′ (δC 158.9). Therefore, its structure was established as the 1'-O-methylated 5 and named otteacumiene F.
By comparing spectroscopic data with literatures, the structures of 35 known compounds were elucidated as (1E, 4E)-1, 7-di(4-methoxyphenyl)-1, 4-heptadiene (7) [15], tedarenes A (8) [16], trans-cinnamic acid (9) [17], p-hydroxymethylcinnamate (10) [18], trans-p-hydroxyl ethyl cinnamate (11) [19 3-(4-hydroxyphenyl)acrylic acid benzyl ester (12) [20], (2E)-3-(4-hydroxyphenyl)-2-propenoic acid 2-phenylethyl ester (13) [21] (E)-cinnamyl-(E)-p-coumarate (14) [22], (E)-cinnamyl-(Z)-p-Coumarate (15) [23], (E)-cinnamyl-(E)-ferulate (16) [23], bupleurumin (17) [24], marginatoxin (18) [25], 3-(3, 4-dimethoxybenzyl)-2-(3, 4-methylenedioxybenzyl) butyrolactone (19) [26], Suchilactone (20) [27], osthol (21) [28], micropubescin (22) [29], 8-(2, 3-dihydroxy-3-methylbutyl)-7-methoxy-2H-1-benzopyran-2-one (23) [29, 30], murraol (24) [31] murrayacaurpin B (25) [32] 5, 6-furanocoumarin (26) [33] xanthotoxin (27) [34], isopimpinellin (28) [35], sakuranetin (29) [36] 7, 8-dihydroxyflavanone (30) [37], 5, 3′-dihydroxy-7, 4′-dimethoxyflavanone (31) [38] pinostrobin (32) [39], tectochrysin (33) [40] 5, 7-dihydroxy-flavone (34) [41] desmethylnobiletin (35) [42] (9Z)-heptadeca-1, 9-diene-4, 6-diyn-3-one (36) [43] (8E)-octadeca-1, 8-diene-4, 6-diyne-3, 10-diol (37) [43] 1H-indole-3-carboxylic acid methyl ester (38) [44] 1H-indole-3-carboxaldehyde (39) [45] vanillin (40) [46] and litseagermacrane (41) [47] respectively.
2.2 α-Glucosidase and PTB1B inhibitory activity
To explore the antidiabetic chemical constituents from O. acuminata var. acuminata, the α-glucosidase and PTP1B of inhibitory activities of diaryheptanoids 1-8 were evaluated. The results revealed that these compounds exhibited different levels of inhibitory activity ranging from 38.29% to 103.55% at 50 μM (Table 3), in which 3-8 with inhibition rates more than 50.0% were screened for their IC50 values (acarbose as a positive control). Interestingly, all these compounds exhibited more potential inhibitory activity with IC50 values of 3.81-26.44 μM (Table 3). Especially, 5 represented most effective inhibitor with 60 times more potent than that of acarbose (228.95 μM), the first-line drug for diabetes treatment. It's notable that these diaryheptanoids exhibited negligible effects on PTP1B (Table S11), suggesting that these active ingredients may serve as selective inhibitors on α-glucosidase.
Inhibitory effects of 1-8 against α-Glucosidasea
2.3 Molecular docking studies
To further explore the potential antidiabetic mechanism of the diarylheptanoids, the molecular docking studies were performed by using PyMol program. Compounds 4, 5, and 8 were selected as representative structures of diarylether, linear, and biaryl types of diaryheptanoids for molecular docking against α-glucosidase. The results showed that all the three types of diaryheptanoids act with different cavities mode with that of acarbose [48] which may have contributed to their substantial α-glucosidase inhibitory activities. The molecular docking study for acarbose against α-glucosidase indicated that this first-line medicine formed six hydrogen bonds with ASP-242 (2.9 Å), SER-242 (3.5 Å), GLN-279 (2.8 Å), ARG-422 (2.8 Å), GLU-411 (2.8 Å), and AGR-315 (2.9 Å), respectively. Notably, the 1′-OH of 5 formed three hydrogen bonds with SEP-241 (2.9 Å, 3.2 Å) and ARG-422 (3.0 Å), which might be the reason for its superior activity in contrast to 4 and 8. Correspondingly, 1-OH and 1′-OH of 4 formed two hydrogen bonds with TRY-158 (2.7 Å, 2.8 Å) and 1-OH of 8 formed a hydrogen bond with MET-70 (3.3 Å). Although 4, 5, and 8 in the docking process tended to combine with the same cavity, the difference in the ability to form hydrogen bonds with amino acid residues might be the reason for their different activities (Fig. 5).
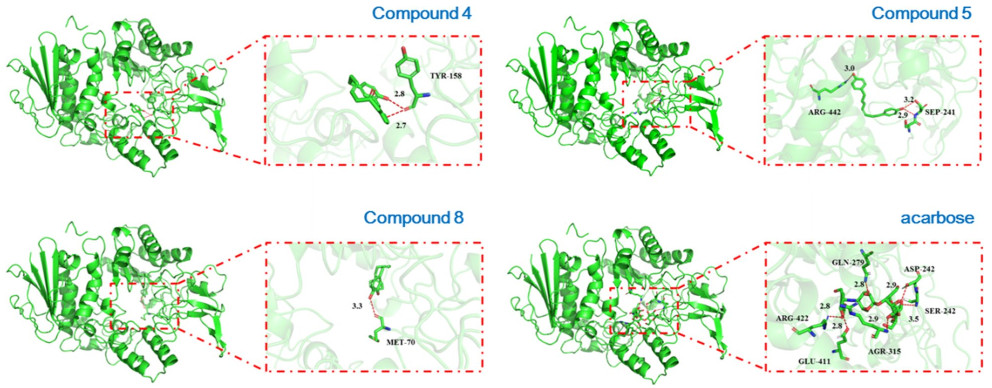
Molecular docking studies of 4, 5, 8, and acarbose against α-glucosidase
3 Conclusion
To the best of our knowledge, this is the first study on the phytochemistry of O. acuminata var. acuminata and their antidiabetic activity. Totally, 41 metabolites ingcluding eight diarylheptanoids (six new ones), eight phenylpropanoids, four lignans, eight coumarins, seven flavonoids, two polyacetylenes, a sesquiterpenoid, two alkaloids, and a vanillin were characterized in this study. Among them, 3 was obtained as a pair of enantiomers whose absolute configurations were determined by calculated ECD method after chiral separation. The biological activity studies displayed that 3-8 exhibited substantial inhibitory activity on α-glucosidase as well as negligible effects on PTP1B, which indicated that these diarylheptanoids might be selective inhibitors of α-glucosidase. Notably, compound 5 was 60-fold stronger than positive control, acarbose. This study implied a common vegetable of Bai, O. acuminata var. acuminata may reduce the incidence of diabetes by inhibiting α-glucosidase. In addition, this work also provides new lead molecules for antidiabetic disease and a reference for medicinal use of O. acuminata var. acuminata.
4 Experimental
4.1 General experimental procedures
IR spectra were measured on a Bruker FT-IR Tensor-27 infrared spectrophotometer with KBr disks. Optical rotations were recorded on a JASCO P-1020 polarimeter. UV spectra were obtained with a Shimadzu UV-2401PC spectrometer. 1D and 2D NMR spectra were performed on a Bruker DRX-600 spectrometer using TMS as an internal standard. The chemical shifts (δ) were expressed in ppm with reference to the solvent signals. HREIMS and HRESIMS analysis were obtained from Waters Xevo TQS and Agilent G6230 TOF mass spectrometers, respectively. Single-crystal X-ray diffraction data were exhibited on a Bruker D8 QUEST diffractometer. Semi-preparative HPLC was performed on a Waters 1525 HPLC with a ZORBAX SB-C18 (9.4 × 250 mm) column. Silica gel (100-200 and 200-300 mesh, Qingdao Marine Chemical Co., Ltd., People's Republic of China), and MCI gel (75-150 μm, Mitsubishi Chemical Corporation, Tokyo, Japan) were used for column chromatography. Fractions were monitored by TLC (GF 254, Qingdao Marine Chemical Co., Ltd.), and spots were visualized by heating silica gel plates immersed in 10% H2SO4 in ethanol. Methanol (HPLC grade) and acetonitrile (HPLC grade) was purchased from CINC High Purity Solvents Co., Ltd (Shanghai, China). 4-Nitrophenyl-α-d-glucopyranoside (PNPG), α-glucosidase, PTP1B assay kit, quercetin, and acarbose were purchased from Sigma Chemical (Merck KGaA, Darmstadt, Germany).
4.2 Plant materials
The whole plants of O. acuminata var. acuminata were collected in Dali Prefecture (Yunnan, China) on October 2019. It was identified by Prof. Yun-Heng Ji in Kunming Institute of Botany, Chinese Academy of Sciences. The specimen of this plant was deposited at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, and the voucher number was KIB L-20191001.
4.3 Extraction and isolation
The dried samples of O. acuminata var. acuminata (20.0 kg) were crushed and extracted four times with methanol for two days each time, and the solvent was recovered under reduced pressure to obtain a crude extract (2.5 kg). The crude extract was eluted with 90% methanol through macroporous resin to obtain 200 g of eluted fractions. The 90% methanol eluent was applied to silica gel column chromatography eluted with dichloromethane (DCM), to afford fraction O-DCM (30.5 g). Fraction O-DMC (30.5 g) was separated over an MCI-gel column (MeOH-H2O from 6:4 to 10:0, v/v) to obtain six fractions from small to large polarities Fr. A-F, which were successively on purified by macroporous resin, silica gel, MCI-gel, RP-C18, and preparative or semi-preparative HPLC chromatographic metheds to give the 41 isolates (detailed process, Fig. S1).
Otteacumiene A (1): Yellow crystals; [α] + 52.4 (c 0.120, MeOH); UV (MeOH) λmax (log ε) 205.0 (4.81), 280.8 (3.50) nm; IR (KBr) νmax 3442, 1632, 1590, 1519, 1503, 1200, 1109, 1030, 983 cm-1; ECD (MeOH) λmax (Δε) 285 (+ 3.75), 271 (- 0.82), 241 (+ 13.52) nm; HRESIMS m/z 293.1182 [M-H]- (calculated for C19H17O3, 293.1178); and 1H and 13C NMR spectroscopic data, Tables 1, 2.
Otteacumiene B (2): Yellow oil; [α] + 384.0 (c 0.098, MeOH); UV (MeOH) λmax (log ε) 206.0 (3.73), 285 (2.57) nm; IR (KBr) νmax 3400, 1630, 1594, 1515, 1503, 1432, 1212, 1135 cm-1; ECD (MeOH) λmax (Δε) 246 (+ 31.34), 200 (- 17.13) nm; HREIMS m/z 294.1262 [M]+ (calculated for C19H18O3, 294.1256); and 1H and 13C NMR spectroscopic data were shown in Tables 1, 2.
Otteacumiene C (3): Colorless crystals; [α] + 1.8 (c 0.075, MeOH); [α] - 68.3 (c 0.048, MeOH) for (-)-3; [α] + 96.8 (c 0.050, MeOH) for (+)-3; UV (MeOH) λmax (logε) 278.5 (3.71), 196.0 (4.83), 258.0 (3.56) nm; IR (KBr) νmax 3408, 1892, 1732, 1516, 1500, 1429, 1208, 1159, 1017 cm-1; ECD (MeOH) λmax (Δε) 285 (+ 19.98), 270 (- 0.21), 259 (+ 2.96), 241 (- 78.41), 224 (+ 30.54), 213 (- 50.23), 204 (+ 1.22) nm for (+)-3; ECD (MeOH) λmax (Δε) 285 (- 13.99), 270 (+ 0.22), 259 (- 2.71), 241 (+ 52.79), 224 (- 22.91), 213 (+ 31, 32), 204 (- 2.74) nm for (-)-3; HRESIMS m/z 349.1407 [M + Na]+ (calculated for C20H22O4Na, 349.1410); and 1H and 13C NMR spectroscopic data were shown in Tables 1, 2.
Otteacumiene D (4): Yellow oil; [α] - 0.8 (c 0.120, MeOH); UV (MeOH) λmax (log ε) 297.5 (4.48), 196.0 (5.09) nm; IR (KBr) νmax 3390, 1703, 1612, 1587, 1508, 1430, 1069, 1053, 994 cm-1; HRESIMS m/z 293.1182 [M-H]- (calculated for C19H17O3, 293.1178); and 1H and 13C NMR spectroscopic data, Tables 1, 2.
Otteacumiene E (5): Green oil; [α] - 0.2 (c 0.098, MeOH); UV (MeOH) λmax (log ε) 200.0 (4.42), 235.4 (4.52), 279.0 (3.69) nm; IR (KBr) νmax 3417, 3017, 2923, 1613, 1514, 1450, 1383, 1246, 1101, 826 cm-1; HRESIMS m/z 279.1390 [M-H]- (calculated for C19H19O2, 279.1385); and 1H and 13C NMR spectroscopic data were shown in Tables 1, 2.
Otteacumiene F (6): Green oil; [α] - 0.6 (c 0.110, MeOH); UV (MeOH) λmax (log ε) 200.2 (4.20), 229.6 (4.24), 277.6 (3.39) nm; IR (KBr) νmax 3425, 2924, 1612, 1513, 1442, 1245, 1177, 1036, 826 cm-1; HRESIMS m/z 293.1547 [M-H]- (calculated for C20H21O2 293.1542); and 1H and 13C NMR spectroscopic data were shown in Tables 1, 2.
Crystal data for 1: C19H18O3, M = 294.33, a = 7.5455(4) Å, b = 9.2259(4) Å, c = 21.7568(10) Å, α = 90°, β = 90°, γ = 90°, V = 1514.58(12) Å3, T = 100.(2) K, space group P212121, Z = 4, μ(Cu Kα) = 0.695 mm-1, 27, 150 reflections measured, 2871 independent reflections (Rint = 0.0429). The final R1 values were 0.0251 (I > 2σ(I)). The final wR(F2) values were 0.0643 (I > 2σ(I)). The final R1 values were 0.0255 (all data). The final wR(F2) values were 0.0645 (all data). The goodness of fit on F2 was 1.060. Flack parameter = 0.08(4). (CDDC: 2156830).
Crystal data for 3: C20H22O4∙CH4O, M = 358.42, a = 10.1053(9) Å, b = 18.5003(16) Å, c = 19.6923(17) Å, α = 90°, β = 90°, γ = 90°, V = 3681.5(6) Å3, T = 100.(2) K, space group Pbca, Z = 8, μ(Cu Kα) = 0.744 mm-1, 60, 171 reflections measured, 3516 independent reflections (Rint = 0.0599). The final R1 values were 0.0366 (I > 2σ(I)). The final wR(F2) values were 0.0943 (I > 2σ(I)). The final R1 values were 0.0386 (all data). The final wR(F2) values were 0.0959 (all data). The goodness of fit on F2 was 1.070. (CDDC: 2155136).
4.4 α-Glucosidase and PTB1B inhibitory activities assay
The α-glucosidase and PTB1B inhibitory activity were conducted according to the previous reports with slight modifications [49, 50]. Briefly, in the α-glucosidase inhibitory assay, after 50 min incubation at 37 ℃, the absorbance value at 405 nm was detected and acarbose was used as positive control. While in the PTB1B inhibitory assay, after addition of phosphate-based detection reagent then incubation at 30 ℃ for 20 min, and absorbance was measured at 620 nm and suramin was used as positive control. The inhibition percentage was calculated as follows: inhibition rate (%) = (E - S)/E × 100% (E is the OD of the control and S is the OD of the sample) and IC50 (50% concentration of inhibition) was calculated by Reed and Muench method.
4.5 Molecular docking studies
To explore the structure-activity relationship, the molecular docking studies was conducted according to the previous reports with slight modifications [51]. In brief, the AutoDock and PyMol software was used to blind docking between 3D structure of α-glucosidase which is downloaded from RCSB PDB website (PDB ID: 3A4A) and compound ligands.
Notes
Acknowledgements
This study was supported financially by the Second Tibetan Plateau Scientific Expedition and Research program (2019QZKK0502) and State Key Laboratory of Phytochemistry and Plant Resources in West China (E0230211Z1 and P2019-ZZ05).
Author contributions
HX Liu carried out the experiments and drafted the manuscript; JZ Ma participated in the experiments; YS Ye performed ECD calculations and revised the manuscript; JJ Zhao completed samples collection; SJ Wan and XY Hu participated in the revision of manuscript; G Xu designed the experiments, revised the manuscript. All authors read and approved the final manuscript.
Declarations
Competing interests
The authors declare no competing interests.
References
-
1.International Diabetes Federation (IDF). IDF Diabetes Atlas, 10th edn. (Brussels, Belgium, 2021). PubMed Google Scholar
-
2.Dahlen AD, Dashi G, Maslov I, Attwood MM, Jonsson J, Trukhan V, Schioth HB, Trends in antidiabetic drug discovery: FDA approved drugs, New Drugs in clinical trials and global sales[J]. Front Pharmacol 12, 807548 (2022) CrossRef PubMed Google Scholar
-
3.Hossain U, Das AK, Ghosh S, Sil PC, An overview on the role of bioactive alpha-glucosidase inhibitors in ameliorating diabetic complications[J]. Food Chem Toxicol 145, 111738 (2020) CrossRef PubMed Google Scholar
-
4.Zhang LH, Chen QY, Li L, Kwong JSW, Jia PL, Zhao PJ, Wang W, Zhou X, Zhang MM, Sun X, Alpha-glucosidase inhibitors and hepatotoxicity in type 2 diabetes: a systematic review and meta-analysis[J]. Sci Rep-Uk 6, 32649 (2016) CrossRef PubMed Google Scholar
-
5.Li YZ, Teng D, Shi XG, Qin GJ, Qin YF, Quan HB, Shi BY, Sun H, Ba JM, Chen B, Du JL, He LJ, Lai XY, Li YB, Chi HY, Liao EY, Liu C, Liu LB, Tang XL, Tong NW, Wang GX, Zhang JA, Wang YM, Xue YM, Yan L, Yang J, Yang LH, Yao YL, Ye Z, Zhang Q, Zhang LH, Zhu J, Zhu M, Ning G, Mu YM, Zhao JJ, Teng WP, Shan ZY, Prevalence of diabetes recorded in mainland China using 2018 diagnostic criteria from the American Diabetes Association: national cross sectional study[J]. BMJ 369, m997 (2020) PubMed Google Scholar
-
6.Mei ZC, Ju HL, Lan L, Long CW, Bo LJ, Li M, Le C, Comparative study of prevalence and influencing factors of diabetes mellitus between Han and Bai ethnic groups in rural areas of Yunnan province[J]. J Kunming Med Univ 42, 33-7 (2021) PubMed Google Scholar
-
7.Zhang Z, Lee C, Gao Z, Li X, Basic research on ancient Bai character recognition based on mobile APP[J]. Wirel Commu Mob Comput 2021, 4059784 (2021) PubMed Google Scholar
-
8.Wu QJ, Textual research on reality and titles of plants. (Beijing: China Publishing House, 1963), p. 443. PubMed Google Scholar
-
9.Wu ZY, Flora Reipublicae Popularis Sinicae. (Beijing: Science Press vol. 8, 1992), p. 160. PubMed Google Scholar
-
10.Tu CY, Lin H, Wang Q, Wu LJ, Yang YL, Zou LJ, Wu QG, The complete chloroplast genome of Ottelia acuminate var. crispa, an endangered aquatic herb with extremely narrow distribution[J]. Mitochondrlal DNA B 6, 1071-2 (2021) CrossRef PubMed Google Scholar
-
11.Lu YH, Tian CR, Gao CY, Wang XY, Yang X, Chen YX, Liu ZZ, Phenolic profile, antioxidant and enzyme inhibitory activities of ottelia acuminata, an endemic plant from southwestern China[J]. Ind Crop Prod 138, 111423 (2019) CrossRef PubMed Google Scholar
-
12.Ebob OT, Babiaka SB, Ntie-Kang F, Natural products as potential lead compounds for drug discovery against SARS-CoV-2[J]. Nat Prod Bioprospect 11, 611-28 (2021) CrossRef PubMed Google Scholar
-
13.Lin Y, Peng XG, Ruan HL, Diarylheptanoids from the fresh pericarps of Juglans hopeiensis[J]. Fitoterapia 136, 104265 (2019) PubMed Google Scholar
-
14.Cheng XL, Li HX, Chen J, Wu P, Xue JH, Zhou ZY, Xia NH, Wei XY, Bioactive diarylheptanoids from alpinia coriandriodora[J]. Nat Prod Bioprospect 11, 63-72 (2021) CrossRef PubMed Google Scholar
-
15.Hoye TR, Ayyad SEN, Beckord HJ, Brown SG, New diarylheptanoids and a hydroxylated ottelione from Ottelia alismoides[J]. Nat Prod Commun 8, 351-8 (2013) PubMed Google Scholar
-
16.Costantino V, Fattorusso E, Mangoni A, Perinu C, Teta R, Panza E, Ianaro A, Tedarenes A and B: structural and stereochemical analysis of two new strained cyclic diarylheptanoids from the marine sponge Tedania ignis[J]. J Org Chem 77, 6377-83 (2012) CrossRef PubMed Google Scholar
-
17.Li XG, Zhang LY, Zhang X, Yang GP, Synthesis of trans-cinnsmic acid catalyzed by porous organic framework solidbase[J]. Spec Petrochem 38, 23-7 (2021) PubMed Google Scholar
-
18.Abd El-kader AM, Mahmoud BK, Hajjar D, Mohamed MFA, Hayallah AM, Abdelmohsen UR, Antiproliferative activity of new pentacyclic triterpene and a saponin from Gladiolus segetum Ker-Gawl corms supported by molecular docking study[J]. Rsc Adv 10, 22730-41 (2020) CrossRef PubMed Google Scholar
-
19.Zhu HL, Wang ZZ, Zheng BZ, Qi Z, Zheng X, Li PY, Wang F, Liu JP, Chemical constituents from berries of Physalis pubescens[J]. Chin Tradit Herb Drugs 47, 732-5 (2016) PubMed Google Scholar
-
20.Bankova VS, Synthesis of natural esters of substituted cinnamic-acids[J]. J Nat Prod 53, 821-4 (1990) CrossRef PubMed Google Scholar
-
21.Uesawa Y, Sakagami H, Okudaira N, Toda K, Takao K, Kagaya H, Sugita Y, Quantitative structure-cytotoxicity relationship of cinnamic acid phenetyl esters[J]. Anticancer Res 38, 817-23 (2018) PubMed Google Scholar
-
22.Lotti C, Piccinelli AL, Arevalo C, Ruiz I, De Castro GMM, De Sa LFR, Tessis AC, Ferreira-Pereira A, Rastrelli L, Constituents of Hondurian Propolis with inhibitory effects on Saccharomyces cerevisiae multidrug resistance protein Pdr5p[J]. J Agric Food Chem 60, 10540-5 (2012) CrossRef PubMed Google Scholar
-
23.Mahajan RP, Patil UK, Patil SL, A facile microwave assisted synthesis and antimicrobial activities of naturally occurring (E)-cinnamyl (E)-cinnamates and (E)-aryl cinnamates[J]. Indian J Chem B 46, 1459-65 (2007) PubMed Google Scholar
-
24.Kim SY, Yun-Choi HS, Platelet anti-aggregating activities of bupleurumin from the aerial parts of Bupleurum falcatum[J]. Arch Pharm Res 30, 561-4 (2007) CrossRef PubMed Google Scholar
-
25.Ashour ML, El-Readi MZ, Tahrani A, Eid SY, Wink M, A novel cytotoxic aryltetraline lactone from Bupleurum marginatum (Apiaceae)[J]. Phytochem Lett 5, 387-92 (2012) CrossRef PubMed Google Scholar
-
26.Lee SH, Ban HS, Kim YP, Kim BK, Cho SH, Ohuchi K, Shin KH, Lignans from Acanthopanax chiisanensis having an inhibitory activity on prostaglandin E-2 production[J]. Phytother Res 19, 103-6 (2005) CrossRef PubMed Google Scholar
-
27.Das B, Rao SP, Srinivas KVNS, Yadav JS, Lignans, biflavones and taxoids from Himalayan Taxus-Baccata[J]. Phytochemistry 38, 715-7 (1995) CrossRef PubMed Google Scholar
-
28.Harkar S, Razdan TK, Waight ES, Steroids, chromone and coumarins from Angelica-Officinalis[J]. Phytochemistry 23, 419-26 (1984) CrossRef PubMed Google Scholar
-
29.Bhan MK, Raj S, Nayar MNS, Handa KL, Isoprenylcoumarins from Boenninghausenia-Albiflora[J]. Phytochemistry 12, 3010-1 (1973) CrossRef PubMed Google Scholar
-
30.Atkinson E, Boyd DR, Grundon MF, Coumarins of Skimmia-Japonica[J]. Phytochemistry 13, 853-5 (1974) PubMed Google Scholar
-
31.Ito C, Furukawa H, Three new coumarins from Murraya-Exotica[J]. Heterocycles 26, 1731-4 (1987) CrossRef PubMed Google Scholar
-
32.Wu TS, Liou MJ, Kuoh CS, Coumarins of the flowers of Murraya-Paniculata[J]. Phytochemistry 28, 293-4 (1989) PubMed Google Scholar
-
33.Aslam M, Ali M, Dayal R, Javed K, Coumarins and a naphthyl labdanoate diarabinoside from the fruits of Peucedanum grande C. B Clarke[J]. Z Naturforsch C 67, 580-6 (2012) PubMed Google Scholar
-
34.Stevenson PC, Simmonds MSJ, Yule MA, Veitch NC, Kite GC, Irwin D, Legg M, Insect antifeedant furanocoumarins from Tetradium daniellii[J]. Phytochemistry 63, 41-6 (2003) PubMed Google Scholar
-
35.Abel G, Erdelmeier C, Meier B, Sticher O, Isopimpinellin, a furocoumarin from Heracleum-Sphondylium with chromosome damaging activity[J]. Planta Med 51, 250-2 (1985) PubMed Google Scholar
-
36.Yamauchi Y, Okuyama T, Ishii T, Okumura T, Ikeya Y, Nishizawa M, Sakuranetin downregulates inducible nitric oxide synthase expression by affecting interleukin-1 receptor and CCAAT/enhancer-binding protein beta[J]. J Nat Med 73, 353-68 (2019) PubMed Google Scholar
-
37.Hahm ER, Park S, Yang CH, 7, 8-Dihydroxyflavanone as an inhibitor for Jun-Fos-DNA complex formation and its cytotoxic effect on cultured human cancer cells[J]. Nat Prod Res 17, 431-6 (2003) PubMed Google Scholar
-
38.Balza F, Jamieson L, Towers GHN, Chemical-constituents of the aerial parts of Artemisia-Dracunculus[J]. J Nat Prod 48, 339-40 (1985) PubMed Google Scholar
-
39.Han MS, Lee IK, Kim YS, Kim JT, Choe KR, Yun BS, Flavonoids from Propolis inhibit DNA single strand breakage by the Fenton reaction[J]. J Korean Soc Appl Bi 53, 512-5 (2010) PubMed Google Scholar
-
40.Shi SH, Zhang CN, Liu AJ, Li H, Bi KS, Jia Y, Isolation and identification of chemical constituents from alpinia oxyphylla[J]. Chin J Exp Tradit Med Formulae 19, 97-100 (2013) PubMed Google Scholar
-
41.Benabderrahmane W, Amrani A, Benaissa O, Lores M, Lamas J, de Miguel T, Benayache F, Benayache S, Chemical constituents, in vitro antioxidant and antimicrobial properties of ethyl acetate extract obtained from Cytisus triflorus l'Her[J]. Nat Prod Res 34, 1586-90 (2020) PubMed Google Scholar
-
42.Jayaprakasha GK, Negi PS, Sikder S, Rao LJ, Sakariah KK, Naturforsch Z, Antibacterial activity of Citrus reticulata peel extracts[J]. Z Naturforsch C 55, 1030-4 (2000) PubMed Google Scholar
-
43.Murata K, Iida D, Ueno Y, Samukawa K, Ishizaka T, Kotake T, Matsuda H, Novel polyacetylene derivatives and their inhibitory activities on acetylcholinesterase obtained from Panax ginseng roots[J]. Nat Med Tokyo Jpn 71, 114-22 (2017) PubMed Google Scholar
-
44.Wei F, Dong WX, Min CH, Guang SL, Ling Y, Feng NS, Chemical analysis of the South China Sea spine body sponge Acanthella cavernosa[J]. J Pharm Pract 34, 138-41 (2016) PubMed Google Scholar
-
45.Gutierrez-Lugo MT, Woldemichael GM, Singh MP, Suarez PA, Maiese WM, Montenegro G, Timmermann BN, Isolation of three new naturally occurring compounds from the culture of Micromonospora sp P1068[J]. Nat Prod Res 19, 645-52 (2005) PubMed Google Scholar
-
46.Vusovich OV, Tchaikovskaya ON, Sokolova IV, Vasil'eva NY, Experimental and quantum-chemical study of electronically excited states of protolytic Isovanillin Species[J]. Russ Phys J 57, 86-94 (2014) PubMed Google Scholar
-
47.Brezani V, Lelakova V, Hassan STS, Berchova-Bimova K, Novy P, Kloucek P, Marsik P, Dall'Acqua S, Hosek J, Smejkal K, Anti-infectivity against Herpes simplex virus and selected microbes and anti-inflammatory activities of compounds isolated from Eucalyptus globulus Labill[J]. Viruses-Basel 10, 306 (2018) PubMed Google Scholar
-
48.Hu GL, Peng XR, Dong D, Nian Y, Gao Y, Wang XY, Hong DF, Qiu MH, New ent-kaurane diterpenes from the roasted arabica coffee beans and molecular docking to alpha-glucosidase[J]. Food Chem 345, 128823 (2021) PubMed Google Scholar
-
49.He XF, Geng CA, Huang XY, Ma YB, Zhang XM, Chen JJ, Chemical constituents from Mentha haplocalyx Briq. (Mentha canadensis L.) and their α-glucosidase inhibitory activities[J]. Nat Prod Bioprospect 9, 223-9 (2019) PubMed Google Scholar
-
50.Xu YS, Feng ZL, Zhang T, Lv P, Cao J, Li D, Peng C, Lin LG, Pimarane diterpenoids from the seeds of Caesalpinia minax as PTP1B inhibitors and insulin sensitizers[J]. Molecules 25, 4674 (2020) PubMed Google Scholar
-
51.Hong DF, Hu GL, Peng XR, Wang XY, Wang YB, Al-Romaima A, Li ZR, Qiu MH, Unusual ent-kaurane diterpenes from the Coffea Cultivar S288 coffee beans and molecular docking to alpha-glucosidase[J]. J Agric Food Chem 70, 615-25 (2022) PubMed Google Scholar
Copyright information
© The Author(s) 2022
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
