


Ypsilandrosides U-Y, five new steroidal saponins from Ypsilandra thibetica

Abstract
Phytochemical reinvestigation on the whole plants of Ypsilandra thibetica obtained four new spirostanol glycosides, named ypsilandrosides U-X (1–4), and one new cholestanol glycoside, named ypsilandroside Y (5). Their structures have been established by extensive spectroscopic data and chemical methods. Among them, compound 4 is a rare spirostanol glycoside which possesses a novel 5(6 → 7) abeo-steroidal aglycone, while compound 1 is a first spirostanol bisdesmoside attached to C-3 and C-12, respectively, isolated from the genus Ypsilandra. The induced platelet aggregation activity of the isolates was tested.Graphical Abstract

Keywords
Ypsilandra thibetica Melanthiaceae Ypsilandrosides U-Y Spirostanol saponins Cholestanol saponins1 Introduction
Ypsilandra (Melanthiaceae) is distributed in southwestern China and Myanmar, which contains 5 species according to the updated classification of the Angiosperm Phylogeny Group IV [1]. Among them, Ypsilandra thibetica has been used in folk medicine for treatment of scrofula, dysuria, edema, uterine bleeding, and traumatic hemorrhage in China by the local people [2, 3]. Our previous investigations discovered twenty eight new steroidal glycosides including nineteen spirostanol saponins, two furostanol saponins, three cholestanol saponins, two pregnane glycosides, and two C22-steroidal lactone glycosides from this species [4-10], some of which showed cytotoxicity [4, 5], antifungal [4, 6], antibacterial [6], anti-HIV-1 activities [7], and so on. For further investigation on the chemical constituents of this herb, four new spirostanol saponins (1‒4) and one new cholestanol saponin (5) (Fig. 1) were obtained and structurally characterized. The current paper reports the isolation, structural elucidation, and the induced platelet aggregation activity of these isolates.
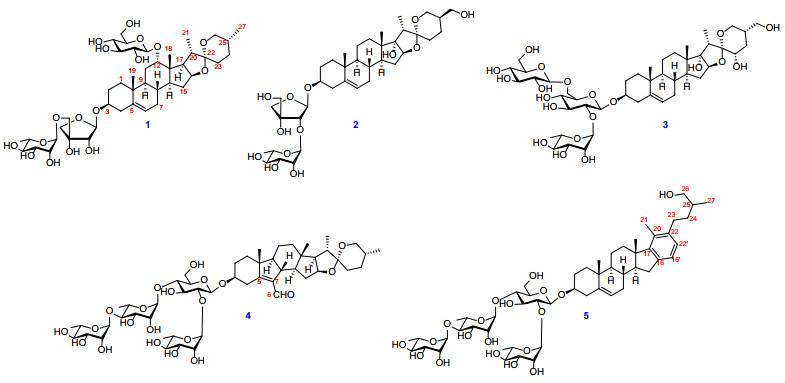
Chemical structures of saponins 1‒5
2 Results and discussion
Compound 1 was isolated as an amorphous powder. Its molecular formula was determined as C44H70O17 by the positive-ion HRESI-MS at m/z 893.4500 [M + Na]+ (calcd. for C44H70O17Na, 893.4505) and 13C NMR data (Table 2). The 1H NMR spectrum of 1 (Table 1) showed four methyl proton signals at δH 0.89 (s, CH3-18), 0.91 (s, CH3-19), 1.38 (d, J = 7.0 Hz, CH3-21), and 0.67 (d, J = 5.4 Hz, CH3-27), one olefinic proton signal at δH 5.18 (o, H-6), while three anomeric protons at δH 5.64 (d, J = 3.2 Hz, H-1'), 5.30 (br s, H-1"), and 4.86 (d, J = 7.8 Hz, H-1"'), which suggested that 1 was a glycoside with three monosaccharide moieties. The 13C NMR spectra displayed 44 carbon signals, of which 17 were assigned to those of one pentose and two hexose units, whereas other 27 ones were assigned to the aglycone moiety, including four methyl groups, nine methylene groups (one oxygenated), ten methine groups (one olefinic and three oxygenated), and four quaternary carbons (one olefinic and one ketal). The above NMR data suggested that compound 1 is a typical C-27 steroidal saponin and its aglycone is heloniogen [11]. This deduction can be confirmed by 2D-NMR spectra. The 1H‒1H COSY correlations revealed that the aglycone for 1 had four structural fragments as shown in (Fig. 2). Furthermore, the key HMBC correlations (Fig. 2) from CH3-18 (δH 0.89) to C-12 (δC 82.4)/C-13 (δC 44.9)/C-14 (δC 44.4)/C-17 (δC 53.1), from CH3-19 (δH 0.91) to C-1 (δC 37.1)/C-5 (δC 141.1)/C-9 (δC 49.0)/C-10 (δC 36.9), from CH3-21 (δH 1.38)/H-20 (δH 2.00)/H-23a (δH 1.77)/H-26a (δH 3.53) to C-22 (δC 109.3) were observed. In addition, the ROESY correlations of H-12 (δH 3.88) with H-18 (δH 0.89) and H-20 (δH 2.00) indicated that the OH-12 was α-oriented (Fig. 3).
1H NMR spectroscopic data of compounds 1–5 (δ in ppm, J in Hz, C5D5N)

1H‒1H COSY and Key HMBC correlations of 1‒5

Key ROESY correlations for the aglycone moieties of 1 and 3
For the sugar part, the pentose was inferred as β-D-apiofuranoside by the 13C NMR signals at δc 108.1 (d, C-1'), 78.4 (d, C-2'), 79.0 (s, C-3'), 74.8 (t, C-4'), and 72.9 (t, C-5') with those of corresponding carbons of α- and β-D-apiofuranoside and α- and β-L-apiofuranoside [12, 13]. And the two hexose units were assigned to be a L-rhamnopyranosyl and a D-glucopyranosyl by their NMR data, the acid hydrolysis of 1, and the HPLC analysis (retention time) of their L-cysteine methyl esters followed by conversion into O-tolyl isothiocyanate derivatives and the authentic samples' derivatives. And the β-configuration of glucopyranosyl was revealed by the coupling constant (3J1, 2 > 7.0 Hz) [14], while the anomeric configuration of rhamnopyranosyl was identified as α-orientated on the basis of the chemical shift values of C-3" (δC 72.9) and C-5" (δC 70.6) with those of corresponding carbons of methyl α- and β-rhamnopyranoside [15]. The sequence of the sugar chain at C-3 of the aglycone was established from the following HMBC corrletions: H-1' (δH 5.64) of Api with C-3 (δC 77.5) of the aglycone, H-1" (δH 5.30) of the Rha with C-5' (δC 72.9) of Api, and H-1"' (δH 4.86) of the Glc with C-12 (δC 82.4) of the aglycone (Fig. 2). Thus, the structure of 1 was elucidated as 12-O-β-D-glucopyranosy-(25R)-spirost-5-en-3β, 12β-diol-3-O-α-L-rhamnopyranosyl-(1 → 5)-β-D-apiofuranoside, and named ypsilandroside U.
Compound 2 was isolated as an amorphous powder with a molecular formula of C38H60O13 determined by the positive-ion HRESI-MS at m/z 747.3921 [M + Na]+, (calcd. for C38H60O13Na, 747.3926) and 13C NMR data (Table 2). Its NMR spectra suggested that 2 is a spirostane saponin with a disaccharide chain. Comparison of the 1H and 13C NMR data of 2 (Tables 1 and 2) with those of ypsiparoside C obtained from the same genus [16] revealed that they shared the same aglycone. The two monosaccharides and their absolute configurations were determined as β-D-apiose and α-L-rhamnose by the same methods with compound 1. The HMBC correlations from H-1' (δH 5.72) to C-3 (δC 77.7), and from H-1" (δH 5.85) of the rhamnopyransyl to C-2' (δC 82.4) established the sequence for 3-O-sugar chain as O-α-L-rhamnopyranosyl-(1 → 2)-β-D-apiofuranoside (Fig. 2). Therefore, the structure of 2 was determined as (25R)-spirost-5-en-3β, 17α, 27-triol-3-O-α-L-rhamnopyranosyl-(1 → 2)-β-D-apiofuranoside, and named ypsilandroside V.
13C NMR spectroscopic data of compounds 1–5 (δ in ppm, C5D5N)
Compound 3 was isolated as an amorphous powder and had a molecular formula of C45H72O20 as determined by the positive-ion HRESI-MS data (m/z 955.4505 [M + Na]+, calcd. for C45H72O20Na, 955.4509) and 13C NMR data (Table 2). Inspection of the NMR spectra (Tables 1 and 2) of 3 revealed that it possessed a spirotanol skeleton with a trisaccharide chain consisting of one rhamnopyranosyl and two glucopyranosyls. Comparing its 1H and 13C NMR data (Tables 1 and 2) with those of trillitschonide S6 [17] indicated that they shared the same aglycone. The α-orientations of OH-23 and CH2OH-25 were supported by the ROESY correlations between H-23 (δH 4.00) and H-20 (δH 3.39)/H-25 (δH 2.29) (Fig. 3). The absolute configurations and the anomeric configurations of monosaccharides were determined by the same methods with the above compounds. The sequence of the sugar chain at C-3 of the aglycone was established by the HMBC correlations from H-1' (δH 4.92) to C-3 (δC 76.8), from H-1" (δH 6.31) to C-2' (δC 77.5), and from H-1"' (δH 5.04) to C-6' (δC 69.9) (Fig. 2). Consequently, the structure of 3 was established as (23S, 25S)-spirost-5-en-3β, 17α, 23, 27-tetraol-3-O-β-D-glucopyranosyl-(1 → 6)-[α-L-rhamnopyranosyl-(1 → 2)]-β-D-glucopyranoside, and named ypsilandroside W.
Compound 4 possessed a molecular formula C51H80O21 determined by the HRESI-MS at m/z 1051.5077 [M + Na]+, (calcd. for C51H80O21Na, 1051.5084) and 13C NMR data (Table 2). The UV spectrum of 4 showed absorption maxima at 254.5 nm, suggesting the presence of a conjugated enal system. When comparing its 1H and 13C NMR data (Tables 1 and 2) with those of ypsilandroside H [10], it was suggested that they shared the same sugar sequence and the similar aglycone, except for the compound 4 has no hydroxyl substituent at the C-17. The above deduction could be verified by the HMBC correlations from H-21 (δH 1.13) and H-18 (δH 0.88) to C-17 (δC 62.4) and 1H‒1H COSY correlations between H-16 (δH 4.59) and H-17 (δH 1.80) (Fig. 2). The HMBC correlations from H-1' (δH 5.02) to C-3 (δC 77.7), from H-1" (δH 6.44) to C-2' (δC 77.9), from H-1"' (δH 5.82) to C-4' (δC 77.7), and from H-1"" (δH 6.28) to C-4"' (δC 80.4) confirmed that compound 3 had the same sequence of 3-O-sugar chain as that of ypsilandroside H (Fig. 2). Thus, the structure of 4 was elucidated as (25R)-B-nor(7)-6-carboxaldehyde-spirost-5(7)-en-3β-ol-3-O-α-L-rhamnopyranosyl-(1 → 4)-α-L-rhamnopyranosyl-(1 → 4)-[α-L-rhamnopyranosyl-(1 → 2)]-β-D-glucopyranoside, and named ypsilandroside X.
The molecular formula of compound 5 was determined as C53H82O19 by the HRESI-MS at m/z 1045.5352 [M + Na]+ (calcd. for C53H82O19Na, 1045.5343) and 13C NMR data (Table 2). Its NMR spectra indicated that compound 5 was a cholestane tetraglycosides containing an aromatic ring. Analysis of the 1H and 13C NMR data (Tables 1 and 2) of 5 suggested that it was similar to that of parispseudoside A [18], and the major difference was the absence of a glucopyranosyl group at OH-26 site. With the assistance of HSQC experiment, 1H and 13C NMR data (Tables 1 and 2) showed four anomeric protons at δH 4.96 (o, H-1'), 6.41 (br s, H-1"), 5.84 (br s, H-1"'), and 6.29 (s, H-1"") and their corresponding anomeric carbons at δC 100.2 (C-1'), 102.1 (C-1"), 102.1 (C-1"'), and 103.2 (C-1""). The sequence of sugar units was consistent with that of compound 4 by HMBC experiment (Fig. 2). As a result, the structure of 5 was assigned as homo-aro-cholest-5-en-3β, 26-diol-3-O-α-L-rhamnopyranosyl-(1 → 4)-α-L-rhamnopyranosyl-(1 → 4)-[α-L-rhamnopyranosyl-(1 → 2)]-β-D-glucopyranoside, and named ypsilandroside Y.
Because the whole plants of Y. thibetica has been used in folk medicine for treatment of uterine bleeding and traumatic hemorrhage in China, the isolated compounds (1–5) were evaluated for their induced platelet aggregation activity and ADP (adenosine diphosphate) was used as a positive control. Unfortunately, the results showed all isolated saponins did not exhibit the inducing platelet aggregation activity at the tested concentration of 100 μM.
3 Experimental section
3.1 General experimental procedures
Optical rotations were measured by a JASCO P-1020 polarimeter (Jasco Corp., Japan). UV spectra were recorded on a Shimadzu UV2401 PC spectrophotometer (Shimadzu Corp., Japan). HRESI-MS was recorded on an Agilent 1290 UPLC/6540 Q-TOF mass spectrometer (Agilent Corp., USA). The NMR experiments were performed on Bruker AVANCE III 500, Avance III-600, and AV 800 spectrometers (Bruker Corp., Switzerland). Silica gel (200–300 mesh, Qingdao Marine Chemical Co., Ltd., People's Republic of China), RP-18 (50 μm, Merck, Germany), and Sephadex LH-20 (Pharmacia, Stockholm, Sweden) were used for column chromatography (CC). An Agilent 1260 system (Agilent Corp., America) with a Zorbax SB-C18 column (5 μm, 9.4 × 250 mm) was used for HPLC separation. TLC was carried out on silica gel HSGF254 plates (Qingdao Marine Chemical Co., China) or RP-18 F254 (Merck, Darmstadt, Germany).
3.2 Plant material
The whole plant materials of Y. thibetica were collected in August 2010 from Zhaotong City, Yunnan Provence, China, and identified by Prof. Xin-Qi Chen, Institute of Botany, Chinese Academy of Sciences, Beijing. A voucher specimen was deposited at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.
3.3 Extraction and isolation
The dried whole plants of Y. thibetica (110 kg) were crushed and extracted three times with 70% EtOH under reflux for a 3 h, 2 h and 2 h. Then, the combined extract was concentrated under reduced pressure. The crude extract (30 kg) was passed through YWD-3F macroporous resin and eluted successively with H2O, 40% EtOH, 75% EtOH, and 95% EtOH, respectively. Evaporated 75% EtOH fraction (crude saponin-rich mixture, 10 kg) was subjected to a silica gel column chromatography (CHCl3–MeOH, 20:1 → 8:2, v/v) to give eleven fractions (Fr. A–Fr. K). Fr. C (560 g) was subjected to a silica gel column chromatography (CHCl3–MeOH, 20:1 → 1:1, v/v) to give 14 fractions (Fr. C-1–Fr. C-14). Fr. C-11 (80 mg) was submitted to Sephadex LH-20 (MeOH) and chromatographically separated on an RP-18 column eluted with MeOH–H2O (40:60 → 70:30, v/v) and purified by preparative HPLC (MeCN–H2O, 40:60 → 50:50, v/v) to afford saponin 2 (tR = 12.8 min, 10 mg). Fr. C-13 (45 g) was submitted to Sephadex LH-20 (MeOH) to give three subfractions (C-13–1–C-13–3). Subsequently, Fr. C-13–1 (150 mg) was further purified by preparative HPLC (MeCN–H2O, 25:75 → 35:65, v/v) to afford saponins 5 (tR = 10.8 min, 7 mg) and 4 (tR = 11.9 min, 12 mg), whereas saponins 3 (tR = 11.1 min, 10 mg) and 1 (tR = 14.8 min, 9 mg) were obtained from Fr. C-13–3 (208 mg) by preparative HPLC (MeCN–H2O, 30:70 → 45:55, v/v).
3.4 Physical and spectroscopic data of new glycosides
3.4.1 Ypsilandroside U (1)
Amorphous solid; [α]D18.6‒55.80 (c 0.20, MeOH); 1H (500 MHz, pyridine d5) and 13C (125 MHz, pyridine d5) NMR data, see Tables 1 and 2; HRESIMS m/z 893.4500 [M + Na]+ (calcd. for C44H70O17Na, 893.4505) (Additional file 1).
3.4.2 Ypsilandroside V (2)
Amorphous solid; [α]D18.6‒190.00 (c 0.12, MeOH); 1H (500 MHz, pyridine-d5) and 13C (125 MHz, pyridine-d5) NMR data, see Tables 1 and 2; HRESIMS m/z 747.3921 ([M + Na]+, calcd. for C38H60O13Na, 747.3926) (Additional file 1).
3.4.3 Ypsilandroside W (3)
Amorphous solid; [α]D20.5‒125.67 (c 0.12, MeOH); 1H (500 MHz, pyridine-d5) and 13C (125 MHz, pyridine-d5) NMR data, see Tables 1 and 2; HRESIMS m/z 955.4505 [M + Na]+ (calcd. for C45H72O20Na, 955.4509) (Additional file 1).
3.4.4 Ypsilandroside X (4)
Amorphous solid; [α]D18.6‒106.40 (c 0.15, MeOH); UV (MeOH) λmax (log ε) 202.5 (3.9), 254.5 (3.9) nm; 1H (500 MHz, pyridine-d5) and 13C (125 MHz, pyridine-d5) NMR data, see Tables 1 and 2; HRESIMS m/z 1051.5077 [M + Na]+ (calcd. for C51H80O21Na, 1051.5084) (Additional file 1).
3.4.5 Ypsilandroside Y (5)
Amorphous solid; [α]D18.6‒48.18 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 203 (4.5) nm; 1H (600 MHz, pyridine-d5) and 13C (150 MHz, pyridine-d5) NMR data, see Tables 1 and 2; HRESIMS m/z 1045.5352 [M + Na]+ (calcd. for C53H82O19Na, 1045.5343) (Additional file 1).
3.5 Acid hydrolysis of compounds 1–5 and determination of the absolute configuration of the sugars by HPLC
Compounds 1‒5 (1.0 mg each) in 6 M CF3COOH (1, 4-dioxane-H2O 1:1, 1.0 mL) were heated at 99 ℃ for 2 h, respectively. The reaction mixture was diluted with H2O (1.0 mL) and then extracted with EtOAc (3 × 2.0 mL). Next, each aqueous layer was evaporated to dryness using rotary evaporation. Each dried residue was dissolved in pyridine (1.0 mL) mixed with L-cysteine methyl ester hydrochloride (1.0 mg) (Aldrich, Japan) and heated at 60 ℃ for 1 h. Then, O-tolyl isothiocyanate (5.0 μL) (Tokyo Chemical Industry Co., Ltd., Japan) was added to the mixture, this being heated at 60 ℃ for 1 h. Each reaction mixture was directly analyzed by reversed phase HPLC following the above procedure. Each reaction mixture was directly analyzed by analytical HPLC on a Poroshell 120 SB-C18 column (100 × 4.6 mm, 2.7 μm, Agilent) using an elution of CH3CN‒H2O (20:75 → 40:60, v/v) at a flow rate of 0.6 mL/min. As a result, the sugars in the test compounds were identified as D-glucose and L-rhamnose, respectively, by comparing their molecular weight and retention time with the standards (tR 13.90 min for D-glucose; tR 17.72 min for L-rhamnose).
3.6 Platelet aggregation assays
Turbidometric measurements of platelet aggregation of the samples were performed in a Chronolog Model 700 Aggregometer (Chronolog Corporation, Havertown, PA, USA) according to Born's method [19, 20]. Rabbit platelet aggregation study was completed within 3.0 h of preparation of platelet-rich plasma (PRP). Immediately after preparation of PRP, 250 μL was incubated in each test tube at 37 ℃ for 5.0 min and then 2.5 μL of compounds (100 μM) were individually added. The changes in absorbance as a result of platelet aggregation were recorded. The extent of aggregation was estimated by the percentage of maximum increase in light transmittance, with the buffer representing 100% transmittance. ADP (adenosine diphosphate) was used as a positive control with a 59.5 ± 6.1% maximal platelet aggregation rate at a concentration of 10 μM. 1% DMSO was used as a blank control with a 2.7 ± 0.6% maximal platelet aggregation. Data counting and analysis was done on SPSS 16.0, with experimental results expressed as mean ± standard error.
4 Conclusion
Phytochemical reinvestigation on the whole plants of Y. thibetica obtained four new spirostanol glycosides, named ypsilandrosides U-X (1–4), and one new cholestanol glycoside, named ypsilandroside Y (5). Their structures have been illustrated by extensive spectroscopic data and chemical methods. Among them, compound 4 is a rare spirostanol glycoside which possesses a novel 5(6 → 7) abeo-steroidal aglycone, while compound 1 is a first spirostanol bisdesmoside attached to C-3 and C-12, respectively, obtained from the Ypsilandra species. This investigation enriched the cognition of the chemical constituents in Y. thibetica. Unfortunately, the bioassay results showed the five new saponins have no the activity of inducing platelet aggregation.
Notes
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (U1802287 and 32000280), the Ten Thousand Talents Plan of Yunnan Province for Industrial Technology Leading Talents, and the State Key Laboratory of Phytochemistry and Plant Resources in West China (P2019-ZZ02).
Author contributions
All authors read and approved the final manuscript.
Declarations
Competing interests
The authors declare that there are no conflicts of interest associated with this work.
References
-
1.Li D-Z. The families and genera of Chinese vascular plants, vol. 1. Beijing: China Science Press; 2020. p. 354–5. PubMed Google Scholar
-
2.College JNM. Dictionary of traditional Chinese materia medica. Shanghai: China Shanghai Scientific and Technological Press; 1977. p. 1841. PubMed Google Scholar
-
3.Yunnan Food and Drug Administration. The Yunnan Chinese materia medica standards, Yi Nationality Medicine (III), Kunming: China Shanghai Yunnan Scientific and Technological Press; 2005. p. 5–6. PubMed Google Scholar
-
4.Xie B-B, Liu H-Y, Ni W, Chen C-X. Ypsilandrosides C-G, five new spirostanol saponins from Ypsilandra thibetica[J]. Steroids 74, 950-5 (2009) CrossRef PubMed Google Scholar
-
5.Liu H-Y, Chen C-X, Lu Y, Yang J-Y, Ni W. Steroidal and pregnane glycosides from Ypsilandra thibetica[J]. Nat Prod Bioprospect 2, 11-5 (2012) CrossRef PubMed Google Scholar
-
6.Xie B-B, Liu H-Y, Ni W, Chen C-X, Lu Y, Wu L, Zheng Q-T. Five new steroidal compounds from Ypsilandra thibetica[J]. Chem Biodivers 3, 1211-8 (2006) CrossRef PubMed Google Scholar
-
7.Xie B-B, Chen C-X, Guo Y-H, Li Y-Y, Liu Y-J, Ni W, Yang L-M, Gong N-B, Zheng Y-T, Wang R-R, Lu Y, Liu H-Y. New 23-spirocholestane derivatives from Ypsilandra thibetica[J]. Planta Med 79, 1063-7 (2013) CrossRef PubMed Google Scholar
-
8.Lu Y, Xie B-B, Chen C-X, Ni W, Hua Y, Liu H-Y. Ypsilactosides A and B, two new C22-steroidal lactone glycosides from Ypsilandra thibetica[J]. Helv Chim Acta 94, 92-7 (2011) CrossRef PubMed Google Scholar
-
9.Si Y-A, Yan H, Ni W, Liu Z-H, Lu T-X, Chen C-X, Liu H-Y. Two new steroidal saponins from Ypsilandra thibetica[J]. Nat Prod Bioprospect 4, 315-8 (2014) CrossRef PubMed Google Scholar
-
10.Lu Y, Chen C-X, Ni W, Hua Y, Liu H-Y. Spirostanol tetraglycosides from Ypsilandra thibetica[J]. Steroids 75, 982-7 (2010) CrossRef PubMed Google Scholar
-
11.Nakano K, Murakami K, Takaishi Y, Tomimatsu T, Nohara T. Studies on the constituents of Heloniopsis orientalis (Thunb.) C. Tanaka[J]. Chem Pharm Bull 37, 116-8 (1989) CrossRef PubMed Google Scholar
-
12.Snyder J-R, Serianni A-S. DL-Apiose substituted with stable isotpoes-synthesis, NMR-spectral analysis, and furanose anomerization[J]. Carbohydr Res 166, 85-99 (1987) CrossRef PubMed Google Scholar
-
13.Kitagawa I, Sakagami M, Hashiuchi F, Zhou J-L, Yoshikawa M, Ren J. Apioglycyrrhizin and araboglycyrrhizin, 2 new sweet oleanene-type triterpene oligoglycosides from the root of Glycyrrhiza inflata[J]. Chem Pharm Bull 37, 551-3 (1989) CrossRef PubMed Google Scholar
-
14.Agrawal P-K. NMR-spectroscopy in the structural elucidation of oligosaccharides and glycosides[J]. Phytochemistry 31, 3307-30 (1992) CrossRef PubMed Google Scholar
-
15.Kasai M-O-R, Asakawa J, Mizutani K, Tanaka O. 13C NMR study of α- and β-anomeric pairs of D-mannopyranosides and L-rhamnopyranosides[J]. Tetrahedron 35, 1427-32 (1979) CrossRef PubMed Google Scholar
-
16.Lu T-X, Shu T, Qin X-J, Ni W, Ji Y-H, Chen Q-R, Khanf A, Zhao Q, Liu H-Y. Spirostanol saponins from Ypsilandra parviflora induce platelet aggregation[J]. Steroids 123, 55-60 (2017) CrossRef PubMed Google Scholar
-
17.Yang Y-J, Pang X, Wang B, Yang J, Chen X-J, Sun X-G, Li Q, Zhang J, Guo B-L, Ma B-P. Steroidal saponins from Trillium tschonoskii rhizomes and their cytotoxicity against HepG2 cells[J]. Steroids 156, 108587 (2020) CrossRef PubMed Google Scholar
-
18.Xiao C-M, Huang J, Zhong X-M, Tan X-Y, Deng P-C. Two new homo-aro-cholestane glycosides and a new cholestane glycoside from the roots and rhizomes of Paris polyphylla var. pseudothibetica[J]. Helv Chim Acta 92, 2587-95 (2009) CrossRef PubMed Google Scholar
-
19.Born G-V-R. Aggregation of blood platelets by adenosine diphosphate and its reversal[J]. Nature 194, 927-9 (1962) PubMed Google Scholar
-
20.Born G-V-R, Cross M-J. The aggregation of blood platelets[J]. J Physiol 168, 178-95 (1963) CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2022
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
