


Recent advances on the synthesis of natural pyrrolizidine alkaloids: alexine, and its stereoisomers

Abstract
Natural products have attracted the interest of the scientific community due to their importance and application. Alexine is a naturally polyhydroxylated pyrrolizidine alkaloid that is broadly found in plant sources and isolated from Alexa leiopetala. The biological properties such as glycosidase inhibitors, anti-virus, and anti-HIV activities, makes it interesting target for synthetical studies. This review reports different approaches and methodologies to the synthesis of alexine, and its stereoisomers as the target compounds in numerous studies.Graphical Abstract
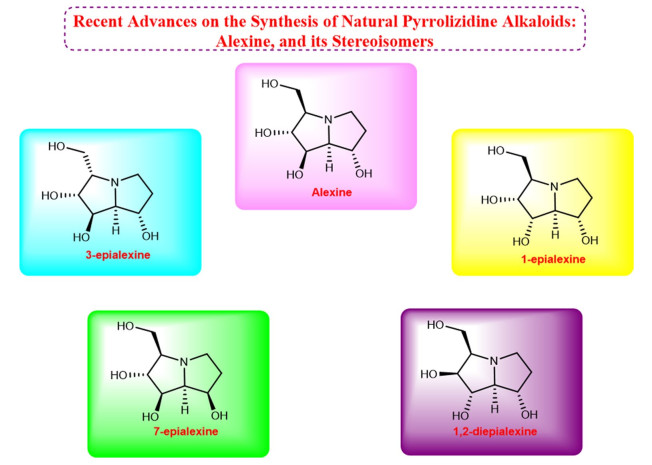
Keywords
Polyhydroxylated pyrrolizidine alkaloids Alexine Natural products Alexa leiopetala1 Introduction
Pyrrolizidine alkaloids are in numerous active natural compounds and isolated mainly from plants of the Asteraceae, Boraginaceae, Fabaceae, and Orchidaceae families [1]. Alexine 1 is a naturally polyhydroxylated pyrrolizidine alkaloid, which was functionalized by the hydroxyl stereocenter groups (Fig. 1). This compound is a class of sugar mimics with the l-aza-bicyclo-[3.3.0]-octane skeleton that they are isolated from plants [2-4].
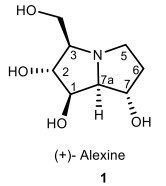
The structures of alexine 1
In 1988, Alexine 1 was isolated from Alexa leiopetala by Nash et al. and its structure and the absolute configuration was revelead using X-ray crystallography [5]. Although many pyrrolizidine alkaloids were previously isolated having carbon substituents at C-1, alexine 1 was the first example of a pyrrolizidine alkaloid with a C-3 hydroxymethyl branch [6].
The potential of these compounds as selective glycosidase inhibitors, as well as their antiviral and retroviral activities, have received much attention from many researchers [7-10]. For example, 7, 7a-diepialexine is one of the useful compounds, which exhibit antiviral effects against HIV virus growth by reducing the cleavage of precursor HIV-1 glycoprotein 160 (gp160) (Fig. 2) [9].
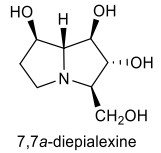
The structure of 7, 7a-diepialexine
Due to the biological activities and stereochemically hydroxyl groups in the pyrrolizidine skeleton, alexine 1 is an important target in organic synthesis. So far, many research groups have effort to synthesis this type of pyrrolizidine alkaloids, and several review articles have been published [11, 12]. Therefore, in continuation of our previous studies about naturally occurring alkaloids [13-21], the synthesis of the natural pyrrolizidine alexine 1 and its stereoisomers were reviewed from 1988 till now with different strategies and methodologies for the synthesis of these compounds.
2 Total synthesis of natural pyrrolizidine alkaloids: alexine, and its Stereoisomers
2.1 The synthetic approach of Fleet and co-workers
In 1988, Fleet et al. [22] reported the synthesis of alexine 1, 3-epialexine 11, and 7-epialexine 13 from α-d-mannofuranoside derivative 3, which was prepared from D-glucose 2 [23]. The diol 3 was reacted with tert-butyldimethylsilyl chloride (TBSCl) to obtain the compound 4, which was esterified with trifluoromethane sulphonic anhydride (Tf2O), followed by hydrogenation over H2/Pd and reaction with benzyl bromide (BnBr) to give the fully protected pyrrolidine 5 in three steps. Deprotection of TBS group through the reaction of resulting product 5 with tetrabutylammonium fluoride (TBAF) provided the compound 6, which was submitted to Swern oxidation [24] [oxalyl chloride (COCl)2/dimethyl sulphoxide (DMSO), triethylamine (Et3N)], followed by reaction with vinyl magnesium bromide (CH2=CHMgBr) to give the epimeric allylic alcohols 7a and 7b. The allylic alcohol 7a was reacted with TBSCl to produce the silyl ether, which was treated with borane dimethylsulfide through the hydroboration reaction, then the resulting silyl ether reacted with alkaline hydrogen peroxide to result in the primary alcohol 9. Then, it was esterified with p-toluenesulphonyl chloride (p-TsCl) to obtain a tosylate, which was cyclized to give the salt 9. Hydrogenolysis of the latter by H2, Pd/C gave the tricyclic compound 10, which was treated with trifluoroacetic acid (TFA), followed by the reduction with NaBH4 to yield the alexine 1. A small amount of 3-epialexine 11 was also obtained and presumably arose from the epimerization of the open-chain form of the intermediate lactol. On the other hand, another alcohol 7b was converted into 7-epialexine 13 by a similar synthetic process (Scheme 1).
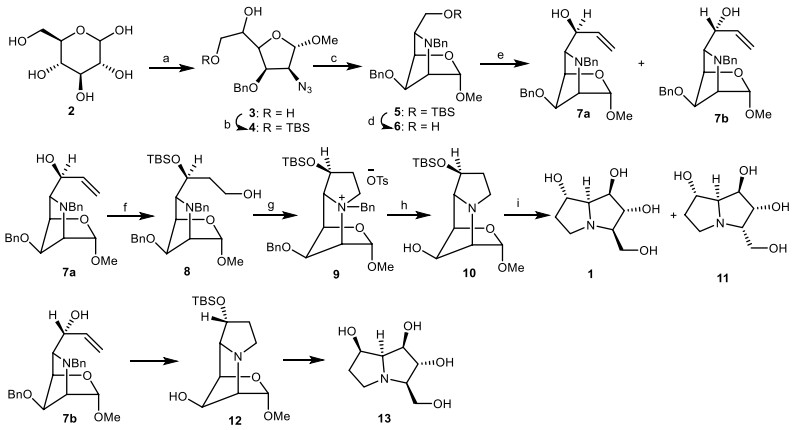
Fleet method for the synthesis of the alexine 1, 3-epialexine 11, and 7-epialexine 13. Reagents and conditions: (a) ref [23]; (b) TBSCl, DMF, 0 ℃, 95%; (c) (i)Tf2O, pyridine, − 30 ℃; (ii) H2, Pd, EtOAc; (iii) BnBr, DMF, NaOH, 77% overall for 3 steps; (d) TBAF, THF, 88%; (e) (i) oxalyl chloride, DMSO, Et3N; (ii) CH2=CHMgBr, THF, 7a: 38%, 7b: 37%; (f) (i) TBSCl, 89%; (ii) BH3.DMS, THF, then H2O2, OH, 67%; (g) (i) p-TsCl, pyridine, CH2Cl2, 77%; (h) H2, Pd/C, aq. acetic acid, 72%; (i) (i) TFA/H2O, 36 h; (ii) NaBH4, EtOH, 54%.
In another study, Fleet et al. investigated the application of the acetonide of heptonolactone 15 as starting material with additional chiral centers, which was derived from L-gluconolactone 14 for the synthesis of 1, 7, 7a-triepialexine 24 and 1, 7a-diepialexine 27 [25, 26]. The acetonide of heptonolactone 15 was converted to azide 16 through the reaction with sodium azide (NaN3). Reduction of azide 16 with diisobutylaluminum hydride (DIBALH) provided the lactol 17, which was reduced by NaBH4 to give diol 18. Then, the diol 18 was protected and mesylated to obtain the mesylate 19. Through the removal of terminal acetonide moiety of mesylate 19, followed by the epoxidation with barium methoxide [Ba(OMe)2], the esterification of the remaining primary alcohol as its triflate, and subsequent treatment with LiCN, the epoxy-nitrile 20 was obtained in four steps. Through the hydrogenolysis of the epoxy-nitrile 20 with H2/Pd black, the key intermediate 21 was obtained. Then, the key intermediate 21 was reacted with ammonium chloride (NH4Cl) to give the bicyclic lactam 22, which was reduced to the amine-borane adduct 23. By the reaction of compound 23 with trifluoroacetic acid (CF3COOH), the target compound 1, 7, 7a-triepialexine 24 was obtained. The bicyclic lactam 22 was oxidized to ketone 25, which was reduced to the alcohol 26. Then, it was converted to 1, 7a-diepialexine 27 through the similar synthetic process, which provided the compound of 1, 7, 7a-triepialexine 24 (Scheme 2).
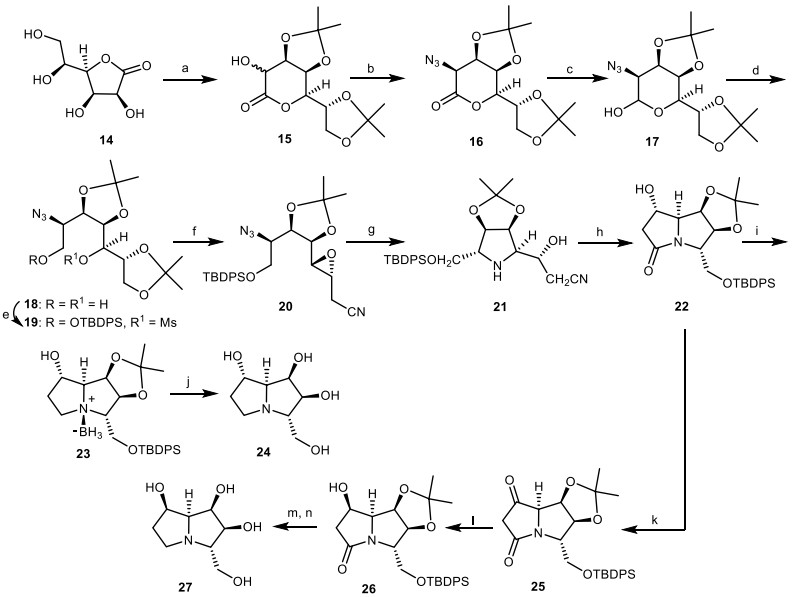
The synthesis of the 1, 7, 7a-triepialexine 24 and 1, 7a-diepialexme 27 by Fleet et al. Reagents and conditions: (a) ref [26]; (b) (i) Tf2O, 85%; (ii) NaN3 in DMF, r.t., 90%; (c) DIBALH in THF; (d) NaBH4 in MeOH; (e) TBDPSCl, imidazole, DMF, 0 ℃, then MsCl, DMAP in pyridine; (f) (i) HOAc in aq. 1, 4-dioxan; (ii) Ba(OMe)2 in MeOH, 0 ℃; (iii) Tf2O in CH2Cl2, − 50 ℃; (iv) LiCN in CH2Cl2/ THF; (g) H2, Pd black, EtOAc; (h) NH4Cl, 35% aq. NH3-EtOH, 100 ℃, 20 h; (i) THF.Borane complex in THF; (j) 50% aq.CF3COOH; (k) PCC, CH2Cl2; (l) NaBH4, EtOH, 0 ℃; (m) THF.Borane complex in THF; (n) 50% aq.CF3COOH.
2.2 The synthetic approach of Ikota and co-workers
Ikota and his co-workers applied optically active pyroglutamic acid derivatives for the natural product synthesis [27-29]. The stereocontrolled synthesis of 1-epialexine 54, 1, 7-diepialexine 55, 1, 7, 7a-triepialexine 24 and 1, 7a-diepialexine 27 were reported based on nonecarbohydrate through (S)-pyroglutamic acid derivative 30 [30, 31]. Dihydroxylation of the unsaturated lactam 29 in the presence of OsO4 as catalyst, followed by isopropylidenation to provide dioxolo derivative 30. The addition of vinylmagnesium bromide (CH2=CHMgBr) to dioxolo derivative 30 through the Grignard reaction provided the α, β-unsaturated ketone 31, which was reduced to diastereometic allylic alcohol 32. Through ozonolysis process of the allylic alcohol 32, followed by the reduction afforded a mixture of diols, which were transformed to the protected ethers 33b and 34b. The resulting ethers were mesylated by methanesulfonyl chloride (MsCl) followed by cyclization in the presence of tert-BuOK to give the protected pyrrolidines 35a and 36a, which were deprotected by tert-butylammonium fluoride (TBAF) to give the alcohols 35b and 36b. On the other hand, 35b also was prepared in an alternative pathway through the removal of lactam moiety of 30, followed by esterification using CH2N2 to provide methyl ester 37. Then, it was reduced to alcohol 38, which was oxidized by Swern reagent followed by treatment with vinylmagnesium bromide to afford the allylic alcohol 32b. The alcohol 32b was mesylated and cyclized to give the 5-vinylpyrrolidine 39. Then, the 5-vinylpyrrolidine 39 converted to pyrrolidine 35b via ozonolysis and subsequent reduction. Swern oxidation of the alcohol 36b, and subsequent reaction with allyl magnesium chloride provided allylic alcohols 40 and 43, which were protected by MOMCl to their methoxymethyl (MOM) ethers, followed by the conversion of N-tert-butyloxycarbonyl (N-Boc) group into the N-Benzyl (N-Bn) protecting group to give the epimeric mixture of 42 and 45. Then, they were ozonolized and reduced to the alcohol 46, which was mesylated and cyclized to the pyrrolizidine skeleton, followed by hydrogenolysis, and then their acidic treatment gave 1, 7a-diepialexine 27 and 1, 7, 7a-triepialexine 24. Through a similar sequence and synthetic pathways, 1-epialexine 54 (α-OH) and 1, 7-diepialexine 55 (β-OH) produced using 35b (Scheme 3).
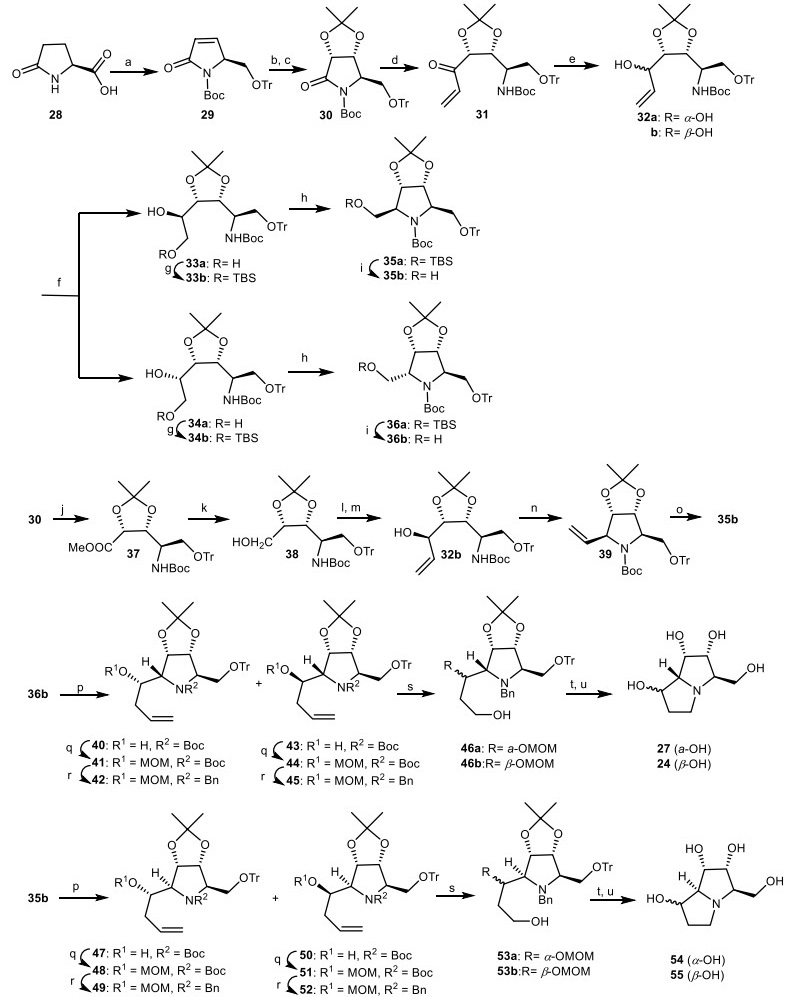
The synthesis of the 1-epialexine 54, 1, 7-diepialexine 55, 1, 7a-diepialexine 27, and 1, 7, 7a-triepialexine 24 by Ikota and his co-workers. Reagents and conditions: (a) ref [28]; (b) OsO4, NMO, acetone/H2O, r.t., 20 h, 88%; (c) (CH3)2C(OCH3)2, p-TsOH, acetone, r.t., 2 h, 88%; (d) CH2=CHMgBr, THF, − 40 ℃, 3 h, 93%; (e) NaBH4, CeCl3.7H2O, MeOH, 0 ℃ to r.t., 10 min to 1 h, 91%; (f) (i) O3, CH2Cl2, -78 ℃, 5 min; (ii) NaBH4, EtOH, 0 ℃, 15 min; (g) TBSCl, imidazole, DMF, 0 ℃, 33b (48%), 34b (19%); (h) (i) MsC1, Et3N, CH2Cl2; (ii) tert-BuOK, THF, 35a (75%), 36a (78%); (i) TBAF, THF, r.t., 35b (78%), 36b (82%); (j) LiOH (aq.), THF/MeOH, then CH2N2, ether; (k) NaBH4, EtOH, 79%; (1) Swern oxidation, − 20 ℃; (m) CH2=CHMgBr, − 78 ℃, 71%; (n) (i) MsC1, Et3N, CH2Cl2; (ii) tert-BuOK, THF; (o) (i) O3, CH2Cl2, -78 ℃; (ii) NaBH4, EtOH, 72%; (p) (i) Swern oxidation, − 78 ℃ to 20 ℃, 10 min; (ii) CH2=CHCH2Li, − 78 ℃, 1 h; (q) MOMCl, N, N-diethylaniline, CH2Cl2, r.t., 40 h; (r) (i) TBSOTf, 2, 6-lutidine, CH2Cl2, r.t., 3 h; (ii) TBAF, THF, (iii) BnBr, K2CO3, acetone, r.t., 2 h; (s) O3, CH2Cl2, − 78 ℃, then NaBH4, EtOH, 15 min; (t) MsCl, Et3N, CH2Cl2, r.t., 16 h, then 10% Pd/C, H2, HCl/EtOH, r.t., 1 h; (u) 10% HCl/MeOH, 70 ℃, 27 (52%), 24, 54, 55 (13–21%).
2.3 The synthetic approach of Yoda and co-workers
Yoda et al. [32] reported the first asymmetric total synthesis of (+)-alexine 1 from the benzyl protected D-arabinofuranose derivative 56 as the chiral pool source and the functionalized homochiral lactam 58. The compound 56 was protected with methoxyphenylmethyl amine (MPMNH2) to provide aminal 57, which was reacted with vinylmagnesium bromide (CH2=CHMgBr) via nucleophilic addition, followed by oxidative reaction with pyridinium chlorochromate (PCC) to yield the functionalized homochiral lactam 58. Dihydroxylation of the double bond of compound 58 provided the aldehyde intermediate 59, which underwent BF3·OEt2-induced allylation to obtain the allyl alcohol 60 in two steps. Deprotection of N-MPM moiety of compound 60, and then its OH group-protection with methoxymethyl chloride (MOMCl) provided compound 61, which was submitted to oxidative cleavage of the terminal alkene, followed by reduction to the corresponding alcohol, and then protection by diphenylsilylchloride (DPSCl) and de-tert-butyldicarbonate [(Boc)2O] to provide the N-protected lactam 62. Then, the addition of vinyl Grignard reagent to compound 62, followed by reduction with NaBH4 afforded the compound 63, which was cyclized under basic conditions after mesylation to give the pyrrolidine derivative 64. Then, it was submitted to oxidative cleavage of alkene moiety, reduction, and protection reaction to provide the fully protected pyrrolidine derivative 65, which was converted to the protected pyrrolizidine core 66 by the replacement of the silyl group with the Ts leaving group, and subsequent removing two MOM and Boc groups. Finally, deprotection of the benzyl groups of compound 66 gave the alexine 1 (Scheme 4).
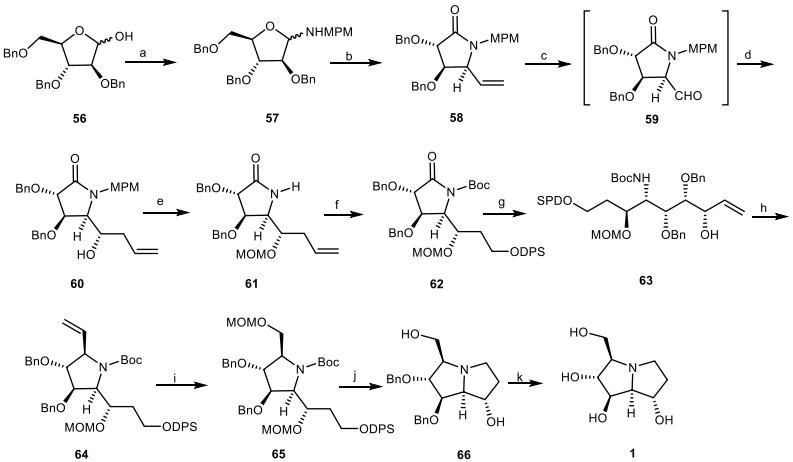
The synthesis of the alexine 1 by Yoda et al. Reagents and conditions: (a) MPMNH2, PhH/CHCl3 (1:1), reflux; (b) (i) CH2=CHMgBr, THF, − 78 to − 40 ℃, 70%; (ii) PCC, CH2Cl2, 68%; (c) (i) OsO4, NMO, acetone/H2O (1:1), 98%; (ii) NaIO4, Et2O/H2O (2:1); (d) allyltrimethylsilane, BF3·OEt2, CH2Cl2, − 78 to − 20 ℃, 82% (two steps); (e) (i) CAN, CH3CN/H2O (9:1), 71%; (ii) MOMCl, i-Pr2NEt, CH2Cl2, 75%; (f) (i) OsO4, NMO, acetone/H2O (1:1), 91%; (ii) NaIO4, Et2O/H2O (2:1); (iii) NaBH4, EtOH, 90%; (iv) DPSCl, imidazole, DMF; (v) (Boc)2O, DMAP, Et3N, CH2Cl2, 99%; (g) (i) CH2=CHMgBr, THF, − 78 ℃; (ii) NaBH4/CeCl3, MeOH, − 45 ℃, 66%; (h) (i) MsCl, Et3N, CH2Cl2; (ii) t-BuOK, THF, 84%; (i) (i) OsO4, NMO, acetone/H2O (1:1), 92%; (ii) NaIO4, Et2O/H2O (2:1); (iii) NaBH4, EtOH, 74%; (iv) MOMCl, i-Pr2NEt, CH2Cl2 99%; (j) (i) Bu4NF, THF.; (ii) p-TsCl, pyridine, 92%; (iii) HCl, MeOH; (iv) K2CO3, MeOH, 94%; (k) H2, 10% Pd: C, EtOH, 70%.
In another study, an efficient pathway for the asymmetric total synthesis of natural (+)-alexine 1 and (−)-7-epi-alexine 13 from protected L-xylose derivative 68 described by Yoda and his co-workers [33]. L-xylose 67 [34] was protected to provide the L-xylose derivative 68, which was treated with 4-methoxybenzylamine (MPM amine), followed by nucleophilic addition of vinyl magnesium chloride to give the amino alcohol 69 as a single isomer. Through the treatment of compound 69 with benzoyl chloride, followed by the reaction with cerium ammonium nitrate (CAN), the MPM group was replaced with the N-benzoyl (Bz) protecting group to provide the amide 70 in two steps. Then, amide 70 was submitted to further protection reactions to obtain the N-tert-butyloxycarbonyl (N-Boc) amide 71, which was deprotected to give the pure NHBoc carbamate 72. The oxidative cleavage of olefinic moiety 72 afforded the aldehyde intermediate, which was treated with vinyl magnesium chloride (CH2=CHMgCl) to obtain allylic alcohol 73a and 73b. Due to the Cram's non-chelation transition structure [35] the carbonyl group was attacked via the less hindered side, so compound 73b is the main product. Its hydroxyl group was protected with methoxymethyl chloride (MOMCl) to give the compound 74, which was subjected to the following reactions of desilylation, mesylation, and t-BuOK-promoted cyclization, leading to the pyrrolidine intermediate 75 in three steps. Hydroboration of the vinyl group of compound 75 gave the primary alcohol 76, which was mesylated, followed by aminocyclization and accompanied by the removal of MOM and Boc group to provide the pyrrolizidine structure 77. By hydrogenolysis of compound 77 with Pd/C, the deprotection of Bn groups was occurred to provide the desired alkaloid 13. The synthesis of (+)-alexine 1 required the inversion in the configuration of the allylic hydroxy center of compound 73b. For this purpose, oxidation of compound 73b with tetra-n-propylammonium perruthenate (TPAP) provided the enone 78, which was reduced diastereoselectively by NaBH4/CeCl3 (Luche reduction) [36], and followed by the protection of the alcohol group to yield allylic alcohol 79. By using the same synthetic strategy to the synthesis of compound 13, the synthesis of (+)-alexine 1 was completed (Scheme 5).
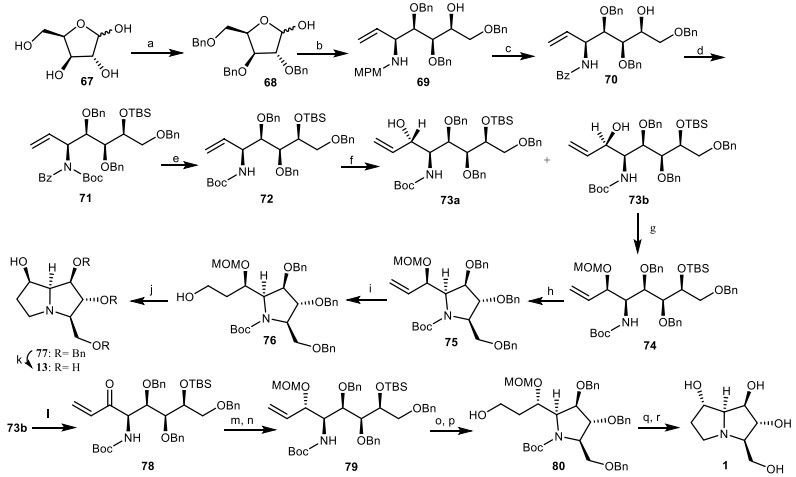
The synthesis of the alexine 1, (−)-7-epi-alexine 13 by Yoda and his co-workers. Reagents and conditions: (a) ref [34]; (b) (i) MPMNH2, toluene, reflux; (ii) CH2=CHMgCl, THF, − 78 ℃ to 0 ℃, 2 h, 75% (two steps); (c) (i) BzCl, CH2Cl2, 89%; (ii) CAN, MeOH, 86%; (d) (i) TBSCl, imidazole, DMF, 89%; (ii) Boc2O, Et3N, DMAP, CH2Cl2, 92%; (e) (Me2N) 2C=NH, reflux, 130 ℃, 8 h, 98%; (f) (i) OsO4, NMO, acetone, 94%; (ii) NaIO4, THF/H2O(1:1); (iii) CH2=CHMgCl, THF, − 78 ℃, 95%; (g) MOMCl, i-Pr2NEt, 98%; (h) (i) Bu4NF, THF, 98%, (ii) MsCl, Et3N, CH2Cl2; (iii) t-BuOK, THF, 84%; (i) (i) 9-BBN, H2O2, NaOH, THF; 94%; (j) (i) MsCl, Et3N, CH2Cl2; (ii) BF3.OEt2, CH2Cl2, − 20 ℃, 84%; (k) Pd/C (10%), HCO2NH4, MeOH, reflux, 2 h, 82%; (l) TPAP, NMO, CH2Cl2, 92%; (m) NaBH4, CeCl3, MeOH, − 45 ℃; 80%; (n) MOMCl, i-Pr2NEt, 96%; (o) (i) Bu4NF, THF, 91%; (ii) MsCl, Et3N, CH2Cl2; (iii) t-BuOK, THF, 87%; (p) 9-BBN, H2O2, NaOH, THF, 97%; (q) (i) MsCl, Et3N, CH2Cl2; (ii) BF3.OEt2, CH2Cl2, − 20 ℃, 84%; (r) Pd/C(10%), HCO2NH4, MeOH; reflux 2 h; 83%.
2.4 The synthetic approach of Pearson and co-workers
Pearson et al. [37, 38] reported the total synthesis of (−)-7-epialexine 13 by reductive amino-cyclization of azidoepoxide as a key step to yield the target compounds. Through the protection reaction, L-xylose 67 was converted to 2, 3, 5-tri-O-benzyl-L-xylofuranose 81 [39], which was transformed to the compound 82 via Wittig olefination. Then, it was converted to the azide 86 via replacement of resultant triflate with the azide group, which the latter was transformed to the aldehyde 84 by ozonolysis process, followed by the Wittig reaction with the silyloxy-substituted ylide 85 to yield the stereoselective Z-alkene 86. It was treated with m-chloroperoxybenzoic acid (m-CPBA) to give epoxides 87a and 87b as a 1:1 mixture of non-separated isomers, which was tosylated to give compounds 88 a/b, followed by the reduction of azido group, and then the aminocyclization of resultant amine to deliver a 2:1 mixture of pyrrolizidines 89, and 90. The target compound, (−)-7-epialexine 14 was produced via the deprotection of 89 using H2, Pd/C (Scheme 6).
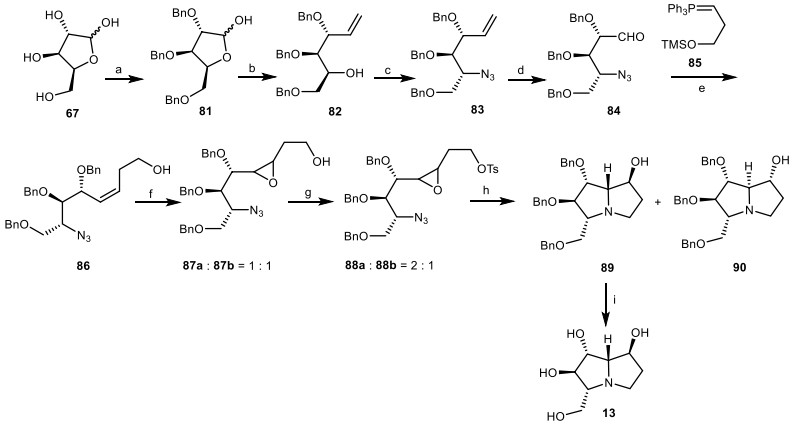
The synthesis of the (−)-7-epialexine 13 by Pearson et al. Reagents and conditions: (a) ref [39]; (b) Ph3PCH3Br, n-BuLi, THF, − 78 ℃ to r.t., 24 h, 66%; (c) (i) Tf2O, pyridine, CH2Cl2, − 40 ℃ to r.t., 3 h; (ii) n-Bu4NN3, PhH, 0 ℃ to r.t., 1 h, 75%; (d) (i) O3, MeOH, CH2Cl2, − 78 ℃, 17 min; (ii) Me2S, − 78 ℃ to r.t., 2.25 h; (e) (i) 85, KN(TMS)2, Me3SiCl, THF, -78 ℃ to r.t., 4 h; (ii) aq. HC1, 35%; (f) m-CPBA, CH2Cl2, 0 ℃ to r.t., 24 h, 65%; (g) TsCl, pyridine, CH2Cl2, − 15 ℃, 48 h, 77%; (h) (i) H2, 10% Pd/C, ether/EtOH (2:1), r.t., 15 h; (ii) K2CO3, EtOH, reflux, 20 h, 71%; (i) H2, 10% Pd/C, EtOH, r.t., 48 h, 87%.
2.5 The synthetic approach of Wong and Romero
Wong and Romero [40], reported the first chemo-enzymatic approach to the synthesis of 7-epialexine 13 without any protecting the group. In the first step, through the Sharpless asymmetric epoxidation, divinylcarbinol 91 was converted to epoxide 92, which was undergoing base-induced rearrangement to give the epoxide 93. Ring-opening of epoxide 93 using ammonia, followed by the protection of resultant amine with ethyl formate provided the formamide 94. Through the ozonolysis, it converted to the mixture of hemiacetals, which were transformed to triol 95 in the presence of allyl bromide and indium. The terminal diol of 95 submitted to NaIO4 cleavage to obtain the corresponding aldehyde, which underwent to aldol reaction with dihydroxyacetone phosphate (DHAP) mediated by fructose-1, 6-diphosphate aldolase (FDPA), followed by the enzymatic cleavage of the phosphate group using acid phosphatase (Pase) to yield tetrol 96, which was in equilibrium with pyranose 97. Ozonolysis of compound 97 under the reductive conditions using H2, Pd/C, followed by acidic cleavage of the formamide, and subsequent reduction of intermediate A provided the target compound 7-epialexine 13 (Scheme 7).
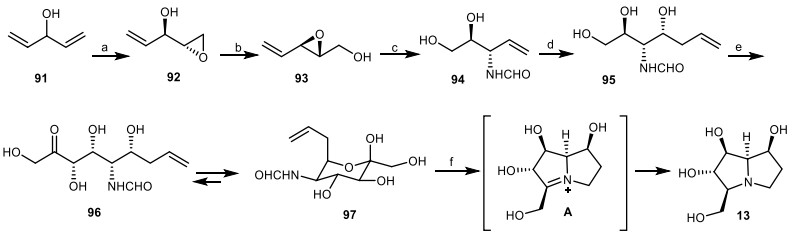
The synthesis of the 7-epialexine 13 by Wong and Romero. Reagents and conditions: (a) (−)-DIPT, C6H5C(CH3)2OOH, Ti(iPrO)4, CH2Cl2, − 35 ℃, 36 h, 86%, > 99% ee; (b) NaOH, r.t., 20 min, 98%; (c) (i) NH4OH, r.t., 24 h; (ii) ethyl formate, EtOH, 90 ℃, 18 h, 95%; (d) (i) O3, MeOH, − 78 ℃; (ii) In, allyl bromide, H2O, r.t., 12 h, 56%; (e) (i) NaIO4, H2O; (ii) DHAP, FDPA, r.t., 72 h; (iii) Pase, 38 ℃, 12 h, 25%; (f) (i) O3, MeOH/H2O, − 78 ℃; (ii) H2, Pd/C, − 78 ℃ to r.t., ; (iii) HCl and H2, Pd/C, r.t., 72 h, 66%.
2.6 The synthetic approach of Donohoe and co-workers
Since the target molecule 13 had the trans-3, 4-diol stereochemistry, the trans-3, 4-diol 101 had to be obtained to enable the synthesis of compound 13. Donohoe's group utilizes N-Boc pyrrole 98 to provide monoacetate 99 [41, 42]. Then, it was submitted to benzyl protection, followed by epoxidation to provide the epoxide derivative 100. Through the ring-opening reaction of epoxide 100, and subsequent deprotection of acetate group under basic conditions gave triol 101. The protection and oxidation of compound 101 under Swern conditions gave aldehyde 102, which was reacted with allyl magnesium bromide to generate the protected secondary alcohol 103. The double bond of compound 103 was cleaved and reduced to the primary hydroxyl group, followed by mesylation with Ms2O to produce 104. Then, it was deprotected and cyclized to obtain pyrrolizidine 105. Removal of protecting groups of compound 105 under acidic hydrogenolysis conditions afforded the (−)-7-epialexine 13 (Scheme 8).
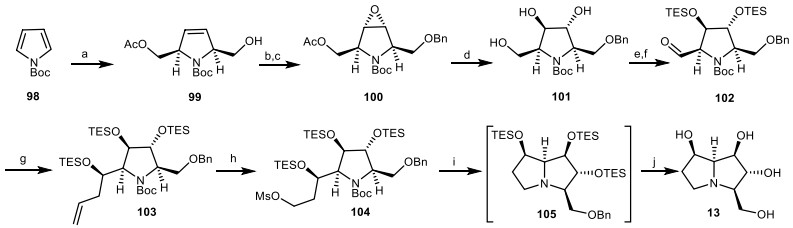
The synthesis of the (−)-7-epi-alexine 13 by Donohoe's group. Reagents and conditions: (a) ref [42]; (b) BnBr, Ag2O, CH2Cl2, r.t., 12 h, 73%; (c) KHSO5, CF3COCH3, EDTA, aq. MeCN, 0 ℃, 30 min, 96% (98%ee); (d) (i) BF3.OEt2, CH2Cl2, − 50 ℃, 14 h; (ii) K2CO3, MeOH, r.t., 2 h, 73%; (e) Et3SiCl, DMAP, Et3N, CH2Cl2, r.t., 16 h, 84%; (f) (COCl)2, DMSO, Et3N, CH2Cl2, − 78 ℃, 1 h, 76%; (g) (i) Allyl magnesium bromide, THF, − 78 ℃, 1 h; (ii) Et3SiCl, DMAP, Et3N, CH2Cl2, r.t., 16 h, 86% (6:1 dr); (h) (i) O3, Ph3P, CH2Cl2, r.t., 12 h, (ii) NaBH4, CH2Cl/MeOH, 0 ℃, 10 min, ; (iii) Ms2O, 2, 6-lutidine, DMAP, CH2Cl2, r.t., 12 h, 74%; (i) TESOTf, 2, 6-lutidine, CH2Cl2, then MeOH, r.t., 12 h; (j) H2, Pd/C, HC1, MeOH, r.t., 89%.
2.7 The synthetic approach of Han and co-workers
Han and his co-workers [43], reported the first asymmetric synthesis of 1, 2-diepialexine 117 from the simple olefin 106 in 10 steps (Scheme 9). The regioselective asymmetric aminohydroxylation of α, β-unsaturated ester 106 afforded the enantioselective syn-amino alcohol 107, followed by the OH protection with BnCl, and the N-acetyl group was transformed to the N-Boc group to provide the carbamate 108, which was reduced to the aldehyde 109. Then, it was reacted with vinyl magnesium bromide to provide the allylic alcohol 110, which was undergone an olefin cross-metathesis [44] with 4-butenol p-tolylsulfonate (C11H14O3S) to yield the trans-olefin 111. It was subjected to VO(acac)2 and produced the mixture of the epoxides 112 and 113, which were treated with HCl, and then by K2CO3 to generate the double cyclized compounds 114 and 115. 1, 2-Diepialexine 116 was produced as a HCl salt, by deprotection of p-methoxybenzyl (PMP) and benzyl group by ceric ammonium nitrate (CAN), and then hydrogenation of compound 115.
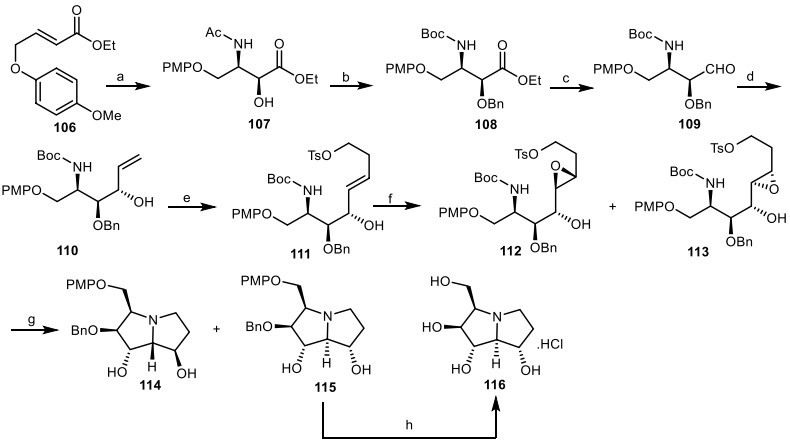
The synthesis of the 1, 2-diepialexine 116 by Han and his co-workers. Reagents and conditions: (a) K2OsO4.2H2O, (DHQD)2-PHAL, LiOH, CH3CONHBr, t-BuOH/H2O (2:1), 4 ℃, 8 h, 70%; (b) (i) NaH, BnCl, DMF, 0 ℃, 10 h, 78%; (ii) (Boc)2O, DMAP, THF, reflux, 4 h, then NH2NH2, MeOH, 4 h, 85%; (c) DIBALH, CH2Cl2, − 78 ℃, 3 h, 90%; (d) CH2=CHMgBr, THF, − 50 ℃, 1 h, then r.t., 1 h, 85%; (e) Grubbs' catalyst, C11H14O3S, CH2Cl2, 65%; (f) VO(acac)2, TBHP, toluene, 74%; (g) (i) HCl, MeOH, (ii) K2CO3, MeOH, 83%; (h) (i) CAN, MeCN/H2O (4:1), 4 ℃; (ii) H2, Pd/C, MeOH, 116 (72%).
2.8 The synthetic approach of Somfai and co-workers
A total asymmetric synthesis of natural alkaloid alexine 1 based on stereoselective [3 + 2] annulation reaction of chiral α-amino aldehyde 119 with 1, 3-bis(silyl)propene 120 was introduced by Somfai et al. [45]. Aldehyde 119 (obtained from α-amino alcohols 117) [46] was reacted with silane 120 (obtained from 1, 3-dichloropropene 118) in the presence of (CH3)3SiCl) [47] to afford pyrrolidine 121, through the stereoselective [3 + 2] annulation reaction. Removal of the silyl group of 121 gave diol 122, which was oxidized chemoselectivity to give aldehyde 123. Then, it was reacted with allyltrimethylsilane (CH2=CHCH2TMS) and TiCl4 to obtain pyrrolidine 124, followed by the protection of OH group to yield the compound 125. The terminal olefin moiety of compound 125 was cleavaged by Lemieux-Johnson oxidation [48] to afford the corresponding aldehyde, which was reduced to alcohol 126 in two steps.The tosyl (Ts) group of compound 126 was removed to provide amine 127. Its hydroxyl group was converted into the corresponding alkylbromide, which was cyclized to the pyrrolizidine 128. Chemoselective oxidation of its silyl group under Tamao-Fleming condition provided pyrrolizidine 129a and N-oxide derivative 130. The acetate group of compound 129a was hydrolyzed to provide the alcohol 129b. The hydrogenolysis of compound 129b and 130 with H2, Pd/C, gave alexine 1 (Scheme 10).
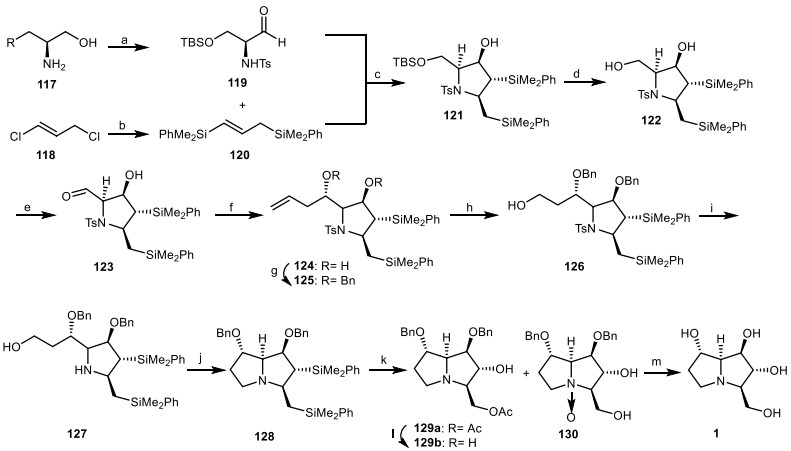
The synthesis of the alexine 1 by Somfai et al. Reagents and conditions: (a) ref [46]; (b) ref [47]; (c) MeAlCl2, CH2Cl2, −78 ℃, 57% (> 40:1 dr); (d) AcOH, THF/H2O, r.t., 96%; (e) TEMPO, NaOCl, KBr, CH2Cl2/H2O, 0 ℃, 99%; (f) CH2=CHCH2TMS, TiCl4, CH2Cl2, − 78 ℃, 1.5 h, 70%; (g) BnBr, KHMDS, THF, − 78 ℃, 85%; (h) (i) OsO4, NaIO4, Py, CH3CN/H2O, r.t., (ii) NaBH4, MeOH, 0 ℃, 72%; (i) NaC10H8, DME, − 60 ℃, 99%; (j) CBr4, Ph3P, Et3N, CH2Cl2, r.t, 64%; (k) Hg(OTf)2, AcO2H, AcOH, r.t., 129a (41%), 129 (21%); (l) LiOH, THF/H2O, r.t., 90%; (m) H2, Pd/C, EtOH, r.t., 70%.
In another work, Somfai and his co-workers [49], developed a new approach based on asymmetric synthesis of the alkaloid alexine 1 through Dynamic Kinetic Resolution/Asymmetric Transfer Hydrogenation Reaction (DKR/ATH) [50]. γ-Alkenyl-β-keto-α-amido esters 131 was subjected to [RuCl2(mesitylene)]2, (S, S)-DPAE, HCO2H/EtN3 through the ATH reaction, using the optimized conditions, furnished diastereo- and enantioselective anti-α-amido-β-hydroxy ester 132 in high yield. Then, it was reduced and protected to give compound 133, which was ozonized, followed by the addition of vinyl magnesium bromide to afford the compound 134. It was reacted with compound 135 via the cross-metathesis reaction, followed by the deprotection of benzyloxymethyl (BOM) group to provide compound 136. Then, it was treated with β-hydroperoxy alcohol 137 through the neighboring group-directed epoxidation in the presence of Ti(OiPr) 4 to give anti epoxide 138, which was deprotected to afford alexine 1 (Scheme 11).
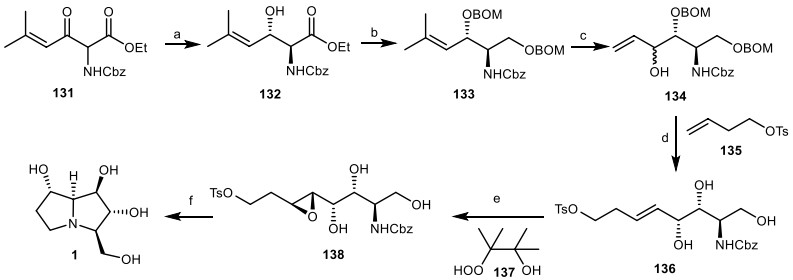
The synthesis of the alexine 1 by Somfai et al. Reagents and conditions: (a) [RuCl2(mesitylene)]2, (S, S)-DPAE, HCO2H/EtN3, dioxane, r.t., 77%; (b) (i) NaBH4, EtOH, r.t.; (ii) BOMCl, i-PrNEt2, CHCl3, r.t., 78%; (c) (i) O3/DMS, CH2Cl2, − 78 ℃ to r.t.; (ii) ZnCl2, CH2=CHMgBr, CH2Cl/CH3Ph, − 78 ℃ to r.t., 58%; (d) (i) Grubbs' catalyst, CH2Cl2, r.t.; (ii) HCI, MeOH, 0 ℃ to r.t., 67%; (e) Ti(OiPr)4, CH2Cl2, 78% (f) H2, Pd/C, MeOH, 76%.
2.9 The synthetic approach of Davies and co-workers
Davis et al. [51] developed an efficient asymmetric synthesis of 1-epialexine 54, based on transannular iodoamination as a critical step. Lithium (R)-N-but-3-enyl-N-(α-methyl-p-methoxybenzyl) amide 141 was reacted with unsaturated ester 140 (derived from D-ribose 139) [52] in the presence of (−)-camphorsulfonyloxaziridine [(−)-CSO] to obtain amino alcohol 142, through the conjugate addition reaction and enolate oxidation. Subsequent treatment of amino alcohol 142 with Grubbs' catalyst gave hexahydroazocine 143, which was reacted with I2, NaHCO3, CH2Cl2 by the diastereoselective transannular iodoamination to give the hydroiodide salt 146, as demonstrated in (Scheme 12). Treatment of compound 146 through the radical-mediated addition of tributyltin hydride (Bu3SnH) in the presence of tetramethylpiperidinyl-N-oxyl (TEMPO) provided a mixture of diastereoisomeric pyrrolizidines 147 and 148. Cleavage of the N–O bond in pyrrolizidine 148 in the presence of Zn, AcOH gave the corresponding hydroxypyrrolizidine 149, which was converted to the triol 319 by reduction of carboxylate group. Oxidative cleavage of triol 150 with NaIO4, followed by reduction with NaBH4 gave the pyrrolizidine alcohol 151. Hydrolysis of acetonide group, through the reaction of pyrrolizidine alcohol 151 with HCl, led to (−)-1-epialexine 54.

The synthesis of the 1-epialexine 54 by Davis et al. Reagents and conditions: (a) ref [52]; (b) (i) THF, − 78 ℃, 2 h; (ii) (−)-CSO, − 78 ℃ to r.t., 12 h, 50% (> 99:1 dr); (c) (i) Grubbs' catalyst, CH2Cl2, 30 ℃, 12 h; (ii) tri(hydroxymethyl) phosphine, Et3N, r.t., 12 h, 73%; (d) I2, NaHCO3, CH2Cl2, r.t., 12 h, 79%; (e) Bu3SnH, TEMPO, PhMe, 70 ℃, 1.5 h, 147 (19%), 148 (69%), (> 99: 1 dr); (f) Zn, AcOH, THF, H2O, 70 ℃, 2 h; (g) LiAlH4, THF, − 78 ℃ to r.t., 12 h; (h) NaIO4, MeOH, H2O, r.t., 4 h then NaBH4, r.t., 12 h; (i) HC1 (3.0 M aq.), MeOH, reflux, 2 h.
2.10 The synthetic approach of Myeong and Ham
In another study, Myeong and Ham achieved the total synthesis of alexine 1 and (−)-7-epi-alexine 13 from anti, syn, anti-oxazine 153 [53]. Treatment of diol 153 (obtained from 152) [54] with 2, 2-dimethylpropanoyl chloride (PivCl) gave compound 154, which was mesylated, followed by oxazine ring cleavage, and simultaneously induced cyclization to afford functionalized pyrrolidine 155. Protection of secondary alcohol 155 by a methoxymethyl (MOM) group gave the compound 156, which was deprotected to give primary alcohol 157. Stereoselective allylation of primary alcohol 157 using MgBr2.OEt2 and BF3.OEt2 resulted in the syn-alcohol 158a and anti-alcohol 158b, which were protected to give compounds 159 and 161. Through the ozonolysis and hydrogenolysis process, they were converted to pyrrolizidine compounds 160 and 162, which were reacted with HCl, followed by neutralization by an ion-exchange resin to provide (−)-7-epi-alexine 13 and alexine 1, respectively (Scheme 13).
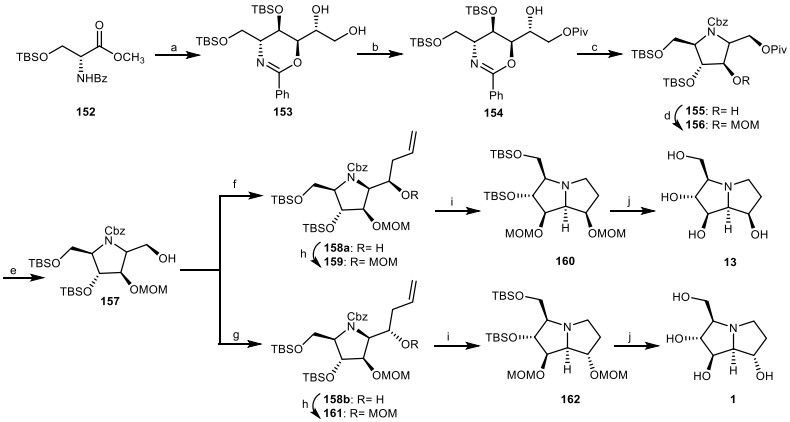
The synthesis of the (−)-7-epi-alexine 13 and alexine 1 by Myeong and Ham. Reagents and conditions: (a) ref [54]; (b) PivCl, pyridine, CH2Cl2, 0 ℃, 1 h, 91%; (c) (i) MsCl, Et3N, CH2Cl2, 0 ℃, 1 h, 93%; (ii) Pd(OH)2/C, H2 (80 psi), r.t., 24 h; (iii) CbzCl, Et3N, CH2Cl2, r.t., 1 h, 66%; (d) MOMCl, DIPEA, DMAP, CH2Cl2, 40 ℃, 24 h, 83%; (e) DIBALH, CH2Cl2, − 78 ℃, 1 h, 85%; (f) MgBr2.OEt2, AllylSnBu3, CH2Cl2, 3 h, 78%; (g) BF3.OEt2, AllylSnBu3, CH2Cl2, 3 h, 73%; (h) MOMCl, DIPEA, DMAP, CH2Cl2, 40 ℃, 24 h, 83–85%; (i) (i) O3, MeOH, −78 ℃, then Me2S; (ii) Pd(OH)2/C, H2, r.t., 24 h, 66–69%; (j) 1 N aq HCl, MeOH, 40 ℃, 3 h, then DOWEX 50X-8 200, 87–90%.
3 Conclusion
Pyrrolizidine alkaloids called alexine 1 being discovered for the first time in 1988. The diverse array of potentially useful biological activities, as well as richness of stereochemical behavior of these alkaloids, have made these compounds attractive and valuable synthetic targets. In the present review, it was tried to highlight the varieties of strategies and methodologies towards the total synthesis of natural alexine 1, and its isomers. It was expected much further development of this type of compounds in synthetic chemistry due to their structural features and biological activities.
Notes
Abbreviations
ATH: Asymmetric transfer hydrogenation; Boc: tert-Butyloxycarbonyl; BOM: Benzyloxymethyl; CAN: Ceric ammonium nitrate; Cbz: Carbobenzyloxy; m-CPBA: meta-Chloroperoxybenzoic acid; CSO: Camphorsulfonyloxaziridine; DHAP: Dihydroxyacetone phosphate; (DHQD)2-AQN: 1, 4-Bis(dihydroquinidinyl) anthraquinone; DHQDPHN: Dihydroquinidine-9-O-(9′-phenanthryl) ether; DIBALH: Diisobutylaluminium hydride; DIPT: Diisopropyl tartrate; DKR: Dynamic kinetic resolution; DPAE: 2-Amino-1, 2-diphenyl-ethanol; DPSCl: Diphenylsilylchloride; DMAP: 4-Dimethylaminopyridine; DMS: Dimethylsulfide; EDTA: Ethylenediaminetetraacetic acid; FDPA: Fructose-1, 6-diphosphate aldolase; KHMDS: Potassium bis(trimethylsilyl)amide; MAPh: Magnesium ammonium phosphate hexahydrate; MOM: Methoxymethyl; MPM: Methoxyphenylmethyl; NMO: N-Methylmorpholine-N-oxide; PCC: Pyridinium chlorochromate; PivCl: Dimethylpropanoyl chloride; TBAF: Tetra-n-butylammonium fluoride; TBDPS: tert-Butyldiphenylsilyl; TBHP: t-Butylhydroperoxide; TBS: tert-Butyldimethylsilyl; TBSOTf: tert-Butyldimethylsilyl trifluoromethanesulfonate; TEMPO: Tetramethylpiperidinyl-N-oxyl; TES: Triethylsilyl; TESOTf: Triethylsilyl trifluoromethanesulfonate; TFA: Trifuoroacetic acid; THF: Tetrahydrofuran; TMS: Trimethylsilyl; TPAP: Tetra-n-propylammonium perruthenate.
Acknowledgements
The authors thank the Research Council of Alzahra University for its support.
Author contributions
GMZ designed and checked the whole manuscript; NJ wrote the manuscript and designed the figures; FM performed the literature review and editing. All authors read and approved the final manuscript.
Declarations
Competing interests
There are no conflicts to declare.
References
-
1.LW Smith, CC Culvenor, Plant sources of hepatotoxic pyrrolizidine alkaloids[J]. J Nat Prod 44, 129-52 (1981) CrossRef PubMed Google Scholar
-
2.Hartmann T, Witte L. Chemistry, biology and chemoecology of the pyrrolizidine alkaloids. In: Pelletier SW, editor. Alkaloids: chemical and biological perspectives, vol. 9. Oxford: Pergamon; 1995. p. 155-233. PubMed Google Scholar
-
3.RJ Nash, LE Fellows, AC Plant, GWJ Fleet, AE Derome, PD Baird, MP Hegarty, AM Scofield, Isolation from castanospermum australe and x-ray crystal structure of 3, 8-diepialexine, (1r, 2r, 3s, 7s, 8r)-3-hydroxymethyl-1, 2, 7-trihydroxypyrrolizidine[(2s, 3r, 4r, 5s, 6r)-2-hydroxymethyl-1-azabicyclo[3.3.0]octan-3, 4, 6-triol][J]. Tetrahedron 44, 5959-64 (1988) CrossRef PubMed Google Scholar
-
4.RJ Nash, LE Fellows, JV Dring, GWJ Fleet, A Girdhar, NG Ramsden, JM Peach, MP Hegarty, AM Scofield, Two alexines[3-hydroxymethyl-1, 2, 7-trihydroxypyrrolizidines] from Castanospermum australe[J]. Phytochemistry 29, 111-4 (1990) CrossRef PubMed Google Scholar
-
5.RJ Nash, LE Fellows, JV Dring, GWJ Fleet, AE Derome, TA Hamor, AM Scofield, DJ Watkin, Isolation from alexa leiopetala and x-ray crystal structure of alexine, (1r, 2r, 3r, 7s, 8s)-3-hydroxymethyl-1, 2, 7-trihydroxypyrrolizidine[(2r, 3r, 4r, 5r, 6s)-2-hydroxymethyl-1 azabicyclo[3.3.0]octan-3, 4, 6-triol], a unique pyrrolizidine alkaloid[J]. Tetrahedron Lett 29, 2487-90 (1988) CrossRef PubMed Google Scholar
-
6.L Jones, J Hollinshead, GW Fleet, AL Thompson, DJ Watkin, ZA Gal, SF Jenkinson, A Kato, RJ Nash, Isolation of the pyrrolizidine alkaloid 1-epialexine from Castanospermum australe[J]. Phytochem Lett 3, 133-5 (2010) CrossRef PubMed Google Scholar
-
7.JE Tropea, RJ Molyneux, G Kaushal, Y Pan, M Mitchell, AD Elbein, Australine, a pyrrolizidine alkaloid that inhibits amyloglucosidase and glycoprotein processing[J]. Biochemistry 28, 2027-34 (1989) CrossRef PubMed Google Scholar
-
8.A Kato, E Kano, I Adachi, RJ Molyneux, AA Watson, RJ Nash, GWJ Fleet, MR Wormald, H Kizu, K Ikeda, N Asano, Australine and related alkaloids: easy structural confirmation by 13CNMR spectral data and biological activities[J]. Tetrahedron Asymmetry 14, 325-31 (2003) CrossRef PubMed Google Scholar
-
9.D Taylor, R Nash, L Fellows, M Kang, A Tyms, Naturally occurring pyrrolizidines: inhibition of α-glucosidase 1 and anti-HIV activity of one stereoisomer[J]. Antivir Chem Chemother 3, 273-7 (1992) CrossRef PubMed Google Scholar
-
10.N Asano, K Ikeda, M Kasahara, Y Arai, H Kizu, Glycosidase-inhibiting pyrrolidines and pyrrolizidines with a long side chain in Scilla peruviana[J]. J Nat Prod 67, 846-50 (2004) CrossRef PubMed Google Scholar
-
11.SG Pyne, M Tang, The structure, biological activities and synthesis of 3-hydroxylpyrrolizidine alkaloids and related compounds[J]. Curr Org Chem 9, 1393-418 (2005) CrossRef PubMed Google Scholar
-
12.J Robertson, K Stevens, Pyrrolizidine alkaloids: occurrence, biology, and chemical synthesis[J]. Nat Prod Rep 34, 62-89 (2017) CrossRef PubMed Google Scholar
-
13.G Mohammadi Ziarani, F Mohajer, R Moradi, P Mofatehnia, The molecular diversity scope of urazole in the synthesis of organic compounds[J]. Curr Org Synth 16, 953-67 (2019) CrossRef PubMed Google Scholar
-
14.G Mohammadi Ziarani, F Mohajer, Z Kheilkordi, Recent progress towards synthesis of the Indolizidine alkaloid 195B[J]. Curr Org Synth 17, 82-90 (2020) CrossRef PubMed Google Scholar
-
15.F Mohajer, G Mohammadi Ziarani, R Moradi, The study of several synthesis methods of Indolizidine (±)-209I and (±)-209B as natural alkaloids[J]. Curr Org Chem 24, 516-35 (2020) CrossRef PubMed Google Scholar
-
16.G Mohammadi Ziarani, F Mohajer, SM Jamali, NA Ebrahim, Quantitative and qualitative bibliometric scope toward the synthesis of rose oxide as a natural product in perfumery[J]. Curr Org Synth 17, 610-24 (2020) CrossRef PubMed Google Scholar
-
17.F Mohajer, G Mohammadi Ziarani, F Taghipour, The multi steps synthetic methods of (±)-Indolizidine 209D as an amphibian natural product in the family of alkaloids[J]. Nat Prod J 11, 448-62 (2020) PubMed Google Scholar
-
18.G Mohammadi Ziarani, R Chênevert, A Badiei, Chemoenzymatic enantioselective formal synthesis of (-)-gephyrotoxin-223[J]. Iran J Chem Chem Eng 25, 31-8 (2006) PubMed Google Scholar
-
19.R Chênevert, G Mohammadi Ziarani, MP Morin, M Dasser, Enzymatic desymmetrization of meso cis-2, 6-and cis, cis-2, 4, 6-substituted piperidines. Chemoenzymatic synthesis of (5S, 9S)-(+)-indolizidine 209D[J]. Tetrahedron Asymmetry 10, 3117-22 (1999) CrossRef PubMed Google Scholar
-
20.R Chênevert, G Mohammadi Ziarani, M Dasser, Chemoenzymatic enantoselective synthesis of (-)-Indolizidine 167 B[J]. Heterocycles 51, 593-8 (1999) CrossRef PubMed Google Scholar
-
21.R Chênevert, G Mohammadi Ziarani, D Caron, M Dasser, Chemoenzymatic enantioselective synthesis of (-)-enterolactone[J]. Can J Chem 77, 223-6 (1999) PubMed Google Scholar
-
22.GW Fleet, M Haraldsson, RJ Nash, LE Fellows, Synthesis from d-glucose of alexine[(1R, 2R, 3R, 7S, 8S)-3-hydroxymethyl-1, 2, 7-trihydroxypyrrolizidine], 3-epialexine and 7-epialexine[J]. Tetrahedron Lett 29, 5441-4 (1988) CrossRef PubMed Google Scholar
-
23.GW Fleet, PW Smith, Methyl 2-azido-3-O-benzyl-2-deoxy-α-dmamnofuranoside as a divergent intermediate for the synthesis of polyhydroxylated piperidines and pyrrolidines: synthesis of 2, 5-dideoxy-2, 5-imino-d-mannitol[2R, 5R-dihydroxymethyl-3R, 4R-dihydroxypyrrolidine][J]. Tetrahedron 43, 971-8 (1987) CrossRef PubMed Google Scholar
-
24.AJ Mancuso, SL Huang, D Swern, Oxidation of long-chain and related alcohols to carbonyls by dimethyl sulfoxide "activated" by oxalyl chloride[J]. J Org Chem 12, 2480-2 (1978) PubMed Google Scholar
-
25.S Choi, I Bruce, A Fairbanks, G Fleet, A Jones, R Nash, L Fellows, Alexines from heptonolactones[J]. Tetrahedron Lett 32, 5517-20 (1991) CrossRef PubMed Google Scholar
-
26.AR Beacham, I Bruce, S Choi, O Doherty, AJ Fairbanks, GW Fleet, BM Skead, JM Peach, J Saunders, DJ Walking, Acetonides of heptonolactones: powerful chirons[J]. Tetrahedron Asymmetry 2, 883-900 (1991) CrossRef PubMed Google Scholar
-
27.N Ikota, A Hanaki, Synthesis of (2S, 3S, 4S)-4-amino-2, 3-dihydroxyhexanedioic acid derivatives from (R)-pyroglutamic acid[J]. Chem Pharm Bull 37, 1087-9 (1989) CrossRef PubMed Google Scholar
-
28.N Ikota, A Hanaki, Synthesis of (-)-Swainsonine and optically active 3, 4-dihydroxy-2-hydroxymethylpyrrolidines[J]. Chem Pharm Bull 35, 2140-3 (1987) CrossRef PubMed Google Scholar
-
29.N Ikota, Synthesis of (2R, 3R, 4R, 5R)-3, 4-dihydroxy-2, 5-dihydroxymethylpyrrolidine and (-)-anisomycin derivative from (S)-pyroglutamic acid derivative[J]. Heterocycles 5, 983-94 (1995) PubMed Google Scholar
-
30.N Ikota, Stereocontrolled synthesis of 1, 7a-diepialexine[J]. Tetrahedron Lett 33, 2553-6 (1992) CrossRef PubMed Google Scholar
-
31.N Ikota, H Nakagawa, S Ohno, K Noguchi, K Okuyama, Stereoselective synthesis of alexine stereoisomers from (S)-pyroglutamic acid[J]. Tetrahedron 54, 8985-98 (1998) CrossRef PubMed Google Scholar
-
32.H Yoda, H Katoh, K Takabe, Asymmetric total synthesis of natural pyrrolizidine alkaloid, (+)-alexine[J]. Tetrahedron Lett 41, 7661-5 (2000) CrossRef PubMed Google Scholar
-
33.M Takahashi, T Maehara, T Sengoku, N Fujita, K Takabe, H Yoda, New asymmetric strategy for the total synthesis of naturally occurring (+)-alexine and (-)-7-epi-alexine[J]. Tetrahedron 64, 5254-61 (2008) CrossRef PubMed Google Scholar
-
34.M Kawana, H Kuzuhara, S Emoto, The use of Grignard reagents in the synthesis of carbohydrates. Ⅲ. The one-way anomerization of methyl glycofuranosides and the opening of their furanose rings[J]. Bull Chem Soc Jpn 54, 1492-504 (1981) CrossRef PubMed Google Scholar
-
35.DJ Cram, FAA Elhafez, Studies in stereochemistry. X. The rule of "Steric control of asymmetric induction" in the syntheses of acyclic systems[J]. J Am Chem Soc 74, 5828-35 (1952) CrossRef PubMed Google Scholar
-
36.JL Luche, Lanthanides in organic chemistry. 1. Selective 1, 2 reductions of conjugated ketones[J]. J Am Chem Soc 100, 2226-7 (1978) CrossRef PubMed Google Scholar
-
37.WH Pearson, JV Hines, A synthesis of (+)-7-epiaustraline and (-)-7-epialexine[J]. Tetrahedron lett 32, 5513-6 (1991) CrossRef PubMed Google Scholar
-
38.WH Pearson, JV Hines, Total syntheses of (+)-australine and (-)-7-epialexine[J]. J Org Chem 65, 5785-93 (2000) CrossRef PubMed Google Scholar
-
39.M MacCoss, A Chen, RL Tolman, Syntheses of all four possible diastereomers of the acyclonucleoside 9-(1, 3, 4-trihydroxy-2-butoxymethyl) guanine from carbohydrate precursors[J]. Tetrahedron Lett 26, 4287-90 (1985) CrossRef PubMed Google Scholar
-
40.A Romero, C-H Wong, Chemo-enzymatic total synthesis of 3-epiaustraline, australine, and 7-epialexine[J]. J Org Chem 65, 8264-8 (2000) CrossRef PubMed Google Scholar
-
41.TJ Donohoe, MD Cheeseman, TJC O'Riordan, JA Kershaw, Synthesis of (+)-DGDP and (-)-7-epialexine[J]. Org Biomol Chem 6, 3896-8 (2008) CrossRef PubMed Google Scholar
-
42.TJ Donohoe, RE Thomas, MD Cheeseman, CL Rigby, G Bhalay, ID Linney, Flexible strategy for the synthesis of pyrrolizidine alkaloids[J]. Org Lett 10, 3615-8 (2008) CrossRef PubMed Google Scholar
-
43.D Chikkanna, OV Singh, SB Kong, H Han, A general asymmetric route for the synthesis of the alexine and australine family of pyrrolizidine alkaloids. The first asymmetric synthesis of 1, 2-diepi-alexine and 1, 2, 7-triepi-australine[J]. Tetrahedron Lett 46, 8865-8 (2005) CrossRef PubMed Google Scholar
-
44.AK Chatterjee, T-L Choi, DP Sanders, RH Grubbs, A general model for selectivity in olefin cross metathesis[J]. J Am Chem Soc 125, 11360-70 (2003) CrossRef PubMed Google Scholar
-
45.M Dressel, P Restorp, P Somfai, Total synthesis of (+)-alexine by utilizing a highly stereoselective[3 + 2] annulation reaction of an N-tosyl-α-amino aldehyde and a 1, 3-bis(silyl)propene[J]. Chem A Eur J 14, 3072-7 (2008) CrossRef PubMed Google Scholar
-
46.J Jurczak, D Gryko, E Kobrzycka, H Gruza, P Prokopowicz, Effective and mild method for preparation of optically active α-amino aldehydes via TEMPO oxidation[J]. Tetrahedron 54, 6051-64 (1998) CrossRef PubMed Google Scholar
-
47.A Guijarro, M Yus, 1, 3-dichloropropene as a source of the 1, 3-dianion derived from propene: synthesis of unsaturated 1, 5-diols and dihydropyrans[J]. Tetrahedron 50, 13269-76 (1994) CrossRef PubMed Google Scholar
-
48.W Yu, Y Mei, Y Kang, Z Hua, Z Jin, Improved procedure for the oxidative cleavage of olefins by OsO4-NaIO4[J]. Org lett 6, 3217-9 (2004) CrossRef PubMed Google Scholar
-
49.L Yu, P Somfai, Enantioselective synthesis of anti-3-alkenyl-2-amido-3-hydroxy esters: application to the total synthesis of (+)-alexine[J]. RSC Adv 9, 2799-802 (2019) CrossRef PubMed Google Scholar
-
50.B Seashore-Ludlow, F Saint-Dizier, P Somfai, Asymmetric transfer hydrogenation coupled with dynamic kinetic resolution in water: synthesis of anti-β-hydroxy-α-amino acid derivatives[J]. Org Lett 14, 6334-7 (2012) CrossRef PubMed Google Scholar
-
51.EA Brock, SG Davies, JA Lee, PM Roberts, JE Thomson, Polyhydroxylated pyrrolizidine alkaloids from transannular iodoaminations: application to the asymmetric syntheses of (-)-hyacinthacine A1, (-)-7a-epi-hyacinthacine A1, (-)-hyacinthacine A2, and (-)-1-epi-alexine[J]. Org Biomol Chem 11, 3187-202 (2013) CrossRef PubMed Google Scholar
-
52.EA Brock, SG Davies, JA Lee, PM Roberts, JE Thomson, Asymmetric synthesis of polyhydroxylated pyrrolizidines via transannular iodoamination with concomitant N-debenzylation[J]. Org Lett 13, 1594-7 (2011) CrossRef PubMed Google Scholar
-
53.I-S Myeong, C Jung, W-H Ham, Total syntheses of (-)-7-epi-alexine and (+)-alexine using stereoselective allylation[J]. Synthesis 51, 3471-6 (2019) CrossRef PubMed Google Scholar
-
54.I-S Myeong, C Jung, J-Y Kim, S-H Park, W-H Ham, Asymmetric total syntheses of (+)-2, 5-dideoxy-2, 5-imino-d-glucitol[(+)-DGDP] and (-)-1-deoxymannojirimycin[(-)-DMJ] via an extended chiral 1, 3-oxazine[J]. Tetrahedron Lett 59, 2422-5 (2018) CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2022
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
