


Clerodane-type Diterpene Glycosides from Dicranopteris pedata
Abstract
Three new clerodane-type diterpene glycosides, (5R, 6S, 8R, 9S, 10R)-6-O-[β-D-glucopyranosyl-(1→4)-α-L-rhamnopyranosyl] cleroda-3, 13(16), 14-diene (1), (5R, 6S, 8R, 9S, 10R, 13S)-6-O-[β-D-glucopyranosyl-(1→ 4)-α-L-rhamnopyranosyl]-2-oxoneocleroda-3, 13-dien-15-ol (2), (5R, 6S, 8R, 9S, 10R)-6-O-[β-D-glucopyranosyl-(1→ 4)-α-L-rhamnopyranosyl]-(13E)-2-oxoneocleroda-3, 14-dien-13-ol (3), together with two known compounds 4 and 5 were isolated from Dicranopteris pedata. The structures of these compounds were elucidated by detailed spectroscopic analysis, and the absolute configuration of compound 2 was determined by ECD calculations. In addition, compound 1 exhibited weak inhibitory activities against SMMC-7721, MCF-7 and SW480.Graphical Abstract
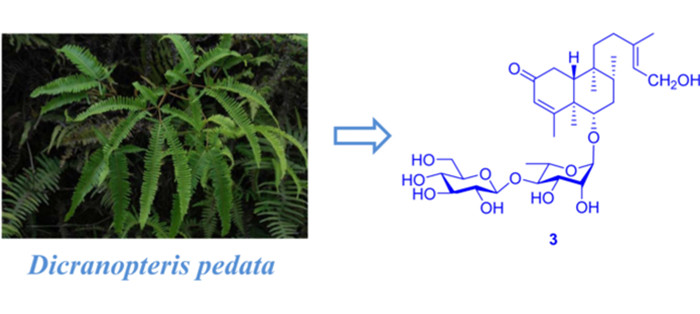
Keywords
Dicranopteris pedata Clerodane-type diterpene glycosides ECD1 Introduction
The genus Dicranopteris is consists of about 10 species, distributed mainly in Asia and widely in Japan, India, Vietnam and China. There are six species in China and they mainly distribute in the south of the Yangtze River such as Yunnan, Sichuan and Guizhou provinces [1]. The whole plant of D. pedata is commonly used as a folk medicine in ancient China for the treatment of hemorrhage, dysentery and empyrosis [2, 3]. Previous researches revealed a diversity of pharmacological properties of the extract of D. pedata, such as antioxidant, antibacterial, antinociceptive, anti-inflammatory and antipyretic activities [4-8]. Up to date, various secondary metabolites including flavonoids [9, 10], phenols [11], proanthocyanidins [12] and clerodane-type diterpene glycosides [10, 13] have been reported from D. pedata. Our previous investigation on this species revealed the presence of two oxygenated phenolic derivatives [14], dichotomains A and B and some clerodane-type diterpene glycosides, in which dichotomain A has exhibited weak anti-HIV activity [15, 16]. As part of our ongoing search for new bioactive metabolites from fern plants, a chemical investigation on the whole plant of D. pedata led to the discovery of three previously undescribed compounds (1–3) (Fig. 1) and two known analogues (compounds 4 and 5) from the acetone extract [15, 16]. This paper describes the details of isolation, structure identification, and cytotoxicity of compounds 1–3.
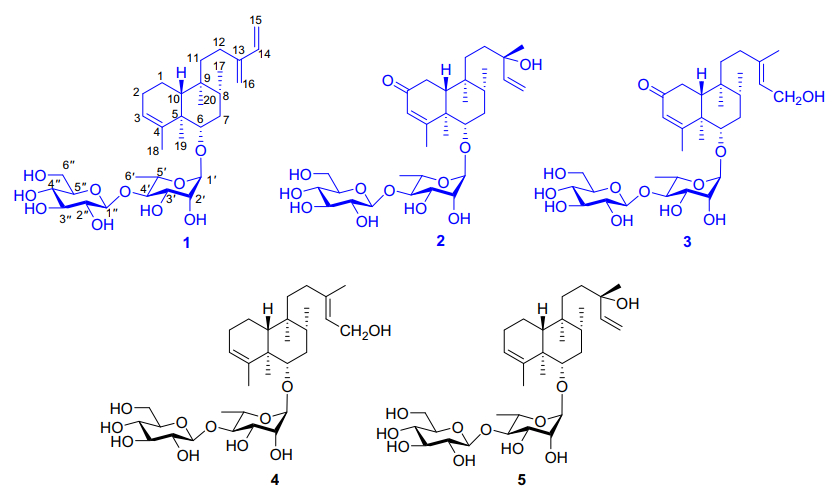
Chemical structures of compounds 1–5
2 Results and Discussion
Compound 1 was isolated as pale yellow powder. Its molecular formula was deduced as C32H52O10 by HRESIMS analysis (found m/z 619.3454 [M + Na]+, calcd for 619.3458), suggesting seven degrees of unsaturation. The 13C and 1H NMR spectra displayed two set of signals ascribed to a diterpene and two hexoses. Five methines (δC 103.2, 72.6, 72.7, 83.5, 68.8) and a methyl (δC 17.9) revealed the presence of rhamnose. In addition, the spectra displayed signals for a glucopyranosyl unit (five methines at δC 105.7, 76.1, 78.1, 71.5, 78.2 and a methylene at δC 62.7). Assignment of each glycosidic proton system was achieved by analysis of 1H-1H COSY, HMBC and HSQC spectra (Fig. 2). The 1H NMR spectrum of the diterpene displayed the presence of four methyls at δH 0.73 (s), 1.08 (s), 0.84 (d, J = 6.7 Hz) and 1.73 (d, J = 1.1 Hz, an olefinic methyl), and six olefinic protons at δH 5.24 (br s), 6.37 (dd, J = 17.6, 10.9 Hz), 5.20 (d, J = 17.6 Hz), 5.04 (d, J = 10.9 Hz), and 4.97 (2H, d, J = 4.4 Hz). The signal at δH 3.43 (dd, J = 11.2, 4.7 Hz) could be attributed to a proton on the oxygenated carbon. Twenty carbons of the diterpene could be classified to four methyls, seven methylenes (two olefinic at δC 113.3, 116.2), five methines (one oxygenated at δC 87.7; two olefinic at δC 123.6, 140.1) and four quaternary carbons (two olefinic at δC 144.6, 148.9). The structure of 1 was identified as a clerodane-type diterpenoid glycoside with two sugar moieties. Aforementioned 1D NMR resonances of 1 showed good agreement with the known compound 5 [16], except for the presence of one olefinic methylene and one olefinic quaternary carbon, together with the absence of a methyl and hydroxyl. The difference of compounds 1 and 5 lie in C-13, C-14, C-15 and C-16. The HMBC correlations (Fig. 2) from H2-16 (δH 4.97) to C-12 (δC 25.5), C-13 (δC 148.9), C-14 (δC 140.1), and H2-15 (δH 5.20, 5.04) to C-13 and C-14, accompanied with the 1H-1H COSY correlations (Fig. 2) between H-14 (δH 6.37) and H2-15 (δH 5.20, 5.04) confirmed the existence of conjugated diene at C-13/C-16 and C-14/C-15. Thus the planar structure of compound 1 was determined.
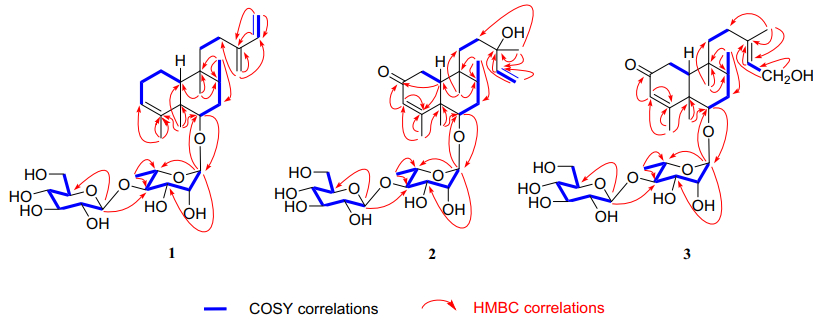
Key 1H–1H COSY and HMBC correlations of compounds 1–3
The relative configurations of α-rhamnopyranosyl and β-glucopyranosyl moiety were identified by the coupling constants of their anomeric protons of the rhamnopyranosyl (H-1ʹ, δH 4.79, J = 1.5 Hz) and glucopyranosyl (H-1ʺ δH 4.59, J = 7.8 Hz), respectively. The location of the sugar chain was determined at C-6 of the aglycone by HMBC correlations (Fig. 2) from the anomeric proton (H-1ʹ, δH 4.79) of the rhamnopyranosyl unit to C-6 (δC 87.7). HMBC correlation from H-1ʺ (δH 4.59, J = 7.8 Hz) to C-4ʹ (δC 83.5) defined a glucopyranosyl (1 → 4) rhamnosyl linkage. The absolute configurations of sugar moieties were determined by the experiments of acid hydrolysis, together with sugar derivatization and analysis with HPLC [16]. The retention times of the product in the test sample are consistent with that of standard sugar derivatives in HPLC (Rha derivative: tR = 6.68 min; Glc derivative: tR = 12.78 min), which gave d-glucose and l-rhamnose.
The relative configuration of 1 was assigned by analysis of its ROESY data (Fig. 4). The correlations of H-6/H-8, and H-6/H-10 indicated that they are spatially close and were thus assigned arbitrarily as β-oriented. Consequently, the correlations of CH3-19/CH3-20 revealed that they are cofacial and adopt a α-orientation. The correlation of H-16/H-14 disclosed the configuration of s-(E)-13(16), 14-diene. The similarity of the NMR results between 1 and 5 suggests the same (5R, 6S, 8R, 9S, 10R) absolute configurations for the aglycone as previously reported. Finally, the structure of 1 was determined as (5R, 6S, 8R, 9S, 10R)-6-O-[β-D-glucopyranosyl-(1 → 4)-α-L-rhamnopyranosyl]cleroda-3, 13(16), 14-diene.
Compound 2 was purified as pale yellow powder with a molecular formula of C32H52O12, which was revealed by the [M + Na]+ ion at m/z 651.3355 (calcd for 651.3357), implying seven indices of hydrogen deficiency. The analysis of 1D NMR spectrum revealed the presence of a diterpene and two hexoses. Each glycosidic proton system was confirmed by analysis of 1H-1H COSY, HMBC and HSQC (Fig. 2). The 1H NMR spectrum of the diterpene displayed the presence of five methyls, one olefinic methylene at δH 5.16 (dd, J = 17.4, 1.5 Hz) and 5.03 (dd, J = 10.8, 1.5 Hz), two olefinic at δH 5.69 (s), 5.84 (dd, J = 17.4, 10.8 Hz). Detailed analysis of its 13C NMR and DEPT spectra revealed 20 carbons resonance which were assigned as five methyls, five methylenes (one olefinic at δC 112.5), five methines (two olefinic at δC 127.1, 146.1) and five quaternary carbons (one olefinic at δC 175.3 and one carbonyl at δC 202.6). The 1H and 13C NMR of 2 were highly analogous to 5 [16], except for the following difference: in the 13C NMR spectrum, the downfield of 2 in chemical shifts of C-3 (δC 127.1) and C-4 (δC 175.3), together with one methylene in 5 was replaced with one carbonyl (δC 202.8). This might be caused by an additional carbonyl at C-2. In the HMBC spectrum, correlations could be observed from δH 5.69 (H-3) to δC 202.8 (C-2), δC 175.3 (C-4) and from δH 1.85 (H-10) to δC 202.8 (C-2), δC 175.3 (C-4). Thus, the carbonyl group was ascertained to be at C-2 position.
The presence of α-l-rhamnose and β-d-glucose were identified by the same method as compound 1. By the HMBC correlations of C-6 (δC 85)/H-1ʹ (δH 4.86) and C-4ʹ (δC 83.3)/H-1ʺ (δH 4.86, d, J = 7.8)(Fig. 2), the linkage positions of the sugar moieties were confirmed. The relative configurations of the diterpene were determined the same for their similar ROESY correlations as 1: H-6/H-10, H-6/H-8, and CH3-19/CH3-20 (Fig. 3). Furthermore, the absolute configuration of 2 was determined by electronic circular dichroism (ECD) experiment which fitted well with the calculated ECD (Fig. 4). Finally, the structure of 2 was determined as (5R, 6S, 8R, 9S, 10R, 13S)-6-O-[β-D-glucopyranosyl-(1 → 4)-α-L-rhamnopyranosyl]-2-ox-oneocleroda-3, 13-dien-15-ol.
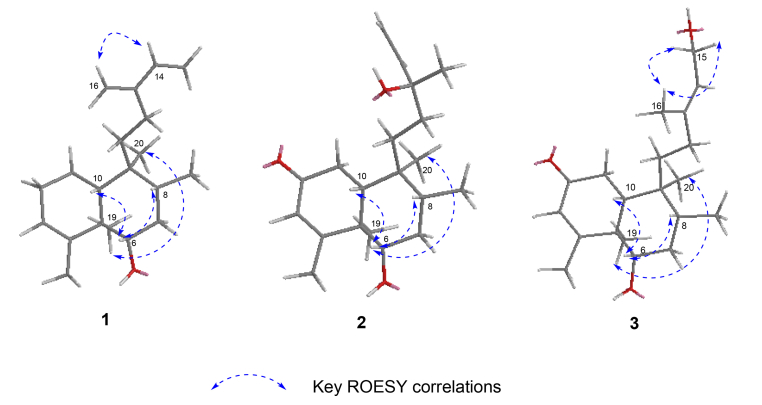
Key ROESY correlations of aglycones of compounds 1–3
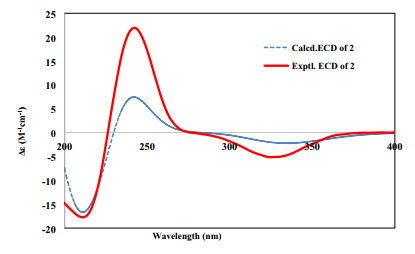
The experimental and calculated ECD spectra of compound 2
Compound 3 was obtained as pale yellow powder. It showed a molecular ion peak at m/z 651.3356 [M + Na]+ (calcd for 651.3351), corresponding to the molecular formula of C32H52O12, which indicating seven degrees of unsaturation. The 1D NMR spectra demonstrated two hexoses and a diterpene. The spectroscopic data of the sugar moieties were consistent with 1 and 2, suggesting the presence of α-l-rhamnopyranosyl moiety and β-D-glucopyranosyl moieties. The diterpene part involved five methyls (one at δC 23.8), five methylenes (one oxygenated at δC 59.1), five methane (one olefinic at δC 125.5) and five quaternary carbons (one olefinic at δC 140.2). Those spectra suggested that the structure of 3 was highly similar to that of 2. In comparison with 2, the chemical shifts of C-14, C-15 and C-16 decreased from δC 146.1, 112.5 and 27.7 to δC 125.5, 59.1 and 23.8 respectively, while C-13 increased from δC 73.8 to 140.2, which testified the position of the double bond of 3 at C-13 and C-14. This was demonstrated again by the HMBC correlations of H2-15/C-13, C-14 and CH3-16/C-13, C-14. In the meantime, C-15 was determined to be a terminal oxymethylene by the chemical shift (δC 59.1) and 1H-1H COSY correlation of H2-15/H-14. Besides, the linkage of the sugar moieties was determined by the identical way with 1 and 2. Thus the plannar structure of compound 3 was determined.
The relative configuration of 3 was assigned by ROESY correlations of H-6/H-10, H-6/H-8, and CH3-19/CH3-20. The ROESY correlations of CH3-16/H2-15 established the E configuration of the C-13/C-14 double bond. The experimental 1D and 2D NMR results of 2 and 3 were similar, which suggesting that they had the same (R, S, R, S, R) absolute configuration at C-5/6/8/9/10, respectively. Finally, the structure of 3 was determined as (5R, 6S, 8R, 9S, 10R)-6-O-[β-D-glucopyranosyl-(1 → 4)-α-L-rhamnopyranosyl]-(13E)-2-oxoneocleroda-3, 14-dien-13-ol.
Additionally, the cytotoxicities of compounds 1–3 were evaluated against five human tumor cell lines HL-60, SMMC-7721, A-549, MCF-7 and SW-480, with cisplatin as the positive control. Compound 1 (40 μM) showed weak inhibitory activities in vitro against SMMC-7721, MCF-7 and SW480 with the inhibition ratio of 48.1%, 48.8% and 39.0%, respectively. However compounds 2 and 3 did not display any activities against the tested cell lines.
3 Conclusion
In conclusion, this research led to the isolation of five compounds including three new and two known clerodane-type diterpene glycosides from D. pedata. Compounds 1–3 were assayed for their cytotoxicitives against five human tumor cell HL-60, SMMC-7721, A-549, MCF-7 and SW-480 with cisplatin as the positive control, in which compound 1 showed weak inhibitory activities in vitro against SMMC-7721, MCF-7 and SW480. This research enriched the chemical diversities of ferns.
4 Experimental Section
4.1 General Experimental Procedures
Optical rotations were carried out on Autopol VI automatoc polarimeter. UV spectra were obtained using a Shimadzu UV-2401 PC spectrophotometer. A Thermo Nicolet iS10 spectrometer was used for measuring IR spectroscopy, which used KBr pellets. 1D and 2D NMR spectra were recorded on Bruker DRX-600 and spectrometers with SiMe4 (TMS) as an internal standard. Chemical shifts (δ) are expressed in ppm with reference to the solvent signals. ESI and HRESIMS were performed on an UPLC-IT-TOF spectrometer. Semi-preparative HPLC was performed on an Agilent 1260 liquid chromatograph with a Zorbax SB-C18 (9.4 mm × 150 mm) column. Column chromatography was carried out using silica gel (100–200 mesh, Qingdao Haiyang Chemical Co. Ltd, Qingdao, People's Republic of China) and macroporous absorption resin (DM 130, Tianjin Haoju Resin Technology Co. Ltd, people's Republic of China). Fractions were monitored by TLC, and spots were visualized by heating silica gel plates sprayed with 10% H2SO4 in EtOH. The reagents used for acid hydrolysis and derivatization are listed: Trifluoroacetic acid (TFA, Beijing Yinuokai Technology Co, Ltd, Beijing, people's Republic of China); 1-Phenyl-3-methyl-5-pyrazolon (PMP, analytical reagent, Tianjin Damao Chemical Reagent Factory); sodium dihydrogen phosphate (NaH2PO4, analytical reagent, Tianjin Damao Chemical Reagent Factory, Tianjin, people's Republic of China); d-glucose and l-rhamnose (Qingdao Jieshikang Biological Technology Co., Ltd, Qingdao, people's Republic of China).
4.2 Plant Material
The fronds of Dicranopteris pedata were collected from Huanglian Mountain, Lvchun County, Yunnan Province, People's Republic of China, in September 2019, identified by Prof. Xiao Cheng (Kunming Institute of Botany, Chinese Academy of Sciences). The voucher specimen (No. 20190921) has been deposited in State Key Laboratory of Phytochemistry and Plant Resource in West China, Kunming Institute of Botany, the Chinese Academy of Sciences.
4.3 Extraction and Isolation of Compounds 1–5
The dry fronds of Dicranopteris pedata (9 kg) were powdered and extracted with acetone (3 × 25 L) for 24 h at room temperature. The acetone extract was concentrated under reduced pressure to yield the residue (700 g), which was then suspended in H2O and subjected to column chromatography over DM 130 eluting with EtOH-H2O (0:10, 5:5, 7:3, 5:95, v/v), EtOH/H2O (5:5, v/v) fraction (230 g) was subjected to silica gel chromatography (CC) using silica gel (200–300 mesh) eluting with CHCl3-MeOH (from 12:1 to 0:1) to yield fractions A-D. Fraction B was subsequently separated on a Sephadex LH-20 CC (MeOH) to give two subfraction Fr.B1 (27 g) and Fr.B2 (25 g). Fr.B1 was divided into ten fractions (from Fr.B1-1 to Fr.B1-10) by using RP-C18 eluting with MeOH-H2O (from 1:9 to 1:1). Fr.B1-10 was separated by CC on silica gel (CHCl3-MeOH, 10:1) and further purified by semi-preparative HPLC with MeCN-H2O (23:77, v/v) to obtain compound 2 (6 mg, tR = 44.11 min). Compound 4 (4 mg, tR = 26.70 min) and 5 (3 mg, tR = 31.35 min) were obtained by MeOH-H2O (40:60, v/v). Compound 3 was separated and purified from Fr.B1-8 by semi-preparative HPLC with MeOH-H2O (48:52, v/v). Fr.B1-9 was separated by CC on silica gel (CHCl3-MeOH, 20:1 to 15:1) and further purified by semi-preparative HPLC with MeOH-H2O (75:25, v/v) to give compound 1 (4 mg, tR = 36.11 min).
4.4 Spectroscopy Data of Compounds 1–3
(5R, 6S, 8R, 9S, 10R)-6-O-[β-D-glucopyranosyl-(1 → 4)-α-L-rhamnopyranosyl]cleroda-3, 13(16), 14-diene (1): pale yellow powder; [α]D26 − 25.8 (c, 0.26, MeOH); UV (MeOH) λmax (log ε): 196.5 (3.26), 208.0 (3.17), 223.5 (3.27); IR (νmax): 3402, 3090, 2956, 2926, 2875 cm−1; HRESIMS at m/z 619.3454 [M + Na]+ (calcd for 619.3458). 1H and 13C NMR data, see Tables 1 and 2.
1H NMR (600 Hz) data (δ in ppm, J in Hz) for compounds 1–3 in CD3OD
13C NMR (150 Hz) data (δ in ppm) for compounds 1–3 in CD3OD
(5R, 6S, 8R, 9S, 10R, 13S)-6-O-[β-D-glucopyranosyl-(1 → 4)-α-L-rhamnopyranosyl]-2-oxoneocleroda-3, 13-dien-15-ol (2): pale yellow powder; [α]26D − 37.98 (c, 0.10, MeOH); UV (MeOH) λmax (log ε): 195.5 (3.13), 209.0 (3.02), 238.5 (3.35); ECD (MeOH) λmax (Δε) 245 (21.24); IR (νmax): 3387, 2964, 2926, 2878, 1652 cm−1; HRESIMS at m/z 651.3355 [M + Na]+ (calcd for 651.3357); 1H and 13C NMR data see Tables 1 and 2.
(5R, 6S, 8R, 9S, 10R)-6-O-[β-D-glucopyranosyl-(1 → 4)-α-L-rhamnopyranosyl]-(13E)-2-oxoneocleroda-3, 14-dien-13-ol (3): pale yellow powder; [α]26D − 18.9 (c, 0.26, MeOH); UV (MeOH) λmax (log ε): 196.0 (3.32), 215.0 (2.99), 238.0 (3.19); IR (νmax): 3404, 2964, 2928, 1716, 1651 cm−1; HRESIMS at m/z 651.3356 [M + Na]+ (calcd for 651.3351). 1H and 13C NMR data see Tables 1 and 2.
4.5 Acid Hydrolysis and Derivatization
Compounds 1–3 were hydrolyzed with trifluoroacetic acid (TFA) by the procedure of the previous reported [17], with minor modifications. Compound 1 (2 mg, 3.35 μmol) was dissolved in 2 M TFA (2 mL), after the reaction mixture was heated to 120 ℃ and stirred for 2 h before cooling to room temperature. Then, extracted with CHCl3 (3 × 1 mL) and the aqueous layer was concentrated for next derivatization. Next, adding 0.3 M NaOH (1 mL) and 0.5 M PMP (1 mL) to the aqueous layer. The reaction was heated to 75 ℃ and stirred for 1.5 h before cooling to room temperature. Then the reaction mixture was quenched with 0.3 M HCl (1 mL) and extracted with CHCl3 (3 × 2 mL). Further analysis the aqueous layer over HPLC [tR = 6.67, 12.78 min; 40% MeOH: 60% sodium phosphate (pH 6.8); 1.5 mL/min]. The acid hydrolysis and HPLC analysis of 2 and 3 were manipulated by the same way as that of 1. Likewise, the standard monosaccharides, d-Glc (2 mg) and l-Rha (2 mg) were derivatized with PMP as compounds 1–3, then HPLC analysis in the same condition as 1–3. The sugar moieties of compounds 1–3 were identified as d-Glc (tR = 12.78 min) and l-Rha (tR = 6.67 min), showing retention times consistent with the standard monosaccharide derivatives.
4.6 Cytotoxicity Assay
MTT assays [18] were used to evaluating the cytotoxicities of compounds 1–3. Five human tumor cell lines including leukemia HL-60, liver cancer SMMC-7721, lung cancer A-549, breast cancer MCF-7 and colon cancer SW-480, were incubated in RMPI-1640 or DMEM medium supplemented with 10% fetal bovine serum and seeded at 3–15 × 103 cells each well of a 96-well cell culture plate. After 12–24 h of incubation at 37 ℃, the tested compound (40 μM, dissolved in DMSO) was added. After incubated for 48 h, each well were added 20 μL MTS [3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt]. Then, they were cultured for further 4 h. The cisplatin was applied as the positive control. The MULTISKAN FC was used to measure the OD value at 492 nm.
Notes
Supplementary Information
The online version contains supplementary material available at https://doi.org/10.1007/s13659-021-00315-y.
Acknowledgements
This work is supported by the Natural Science Foundation of Yunnan province. (No. 202001AT070052)
Data Availability
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits to use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author (s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Declarations
Conflict of interest
All authors declare no conflict of interest.
References
-
1.Editorial Committee of the Flora of China of Chinese Academy of Science, Flora of China, Vol. 2 (3). (Beijing: Science Press, 1980), pp. 118-119. PubMed Google Scholar
-
2.Editorial Board of China Herbal, State Administration of Traditional Chinese Medicine, China Herbal, Vol. 4 (2). (Shanghai: Scientific and Technical Publishers, 1999), pp. 86-87. PubMed Google Scholar
-
3.Q.J. Li, Z.H. Ni, Y. Yang, X.J. Hao, X.S. Yang, Chin. Pharm. J. 51, 1274-1277 (2016) PubMed Google Scholar
-
4.L.J. Ding, Z.H. Zhou, Y.R. Lin, Food Science 26, 77-82 (2005) PubMed Google Scholar
-
5.Y.C. Su, Subtrop. Plant. Sci. 34, 43-45 (2005) PubMed Google Scholar
-
6.Z.A. Zakaria, Z.D.F.A. Ghani, R.N.S.R.M. Nor, H.K. Gopalan, M.R. Sulaiman, A.M.M. Jais, M.N. Somchit, A.A. Kader, J. Ripin, J. Nat. Med. 62, 179-187 (2008) CrossRef PubMed Google Scholar
-
7.Z.A. Zakaria, Z.D.F. Abdul Ghani, R.N.S.R.M. Nor, H.K. Gopalan, M.R. Sulaiman, F.C. Abdullah, Yakugaku Zasshi 126, 1197-1203 (2006) CrossRef PubMed Google Scholar
-
8.Z.A. Zakaria, F.H. Kamisan, M.H. Omar, N.D. Mahmood, F. Othman, S.S. Abdul Hamid, M.N.H. Abdullah, BMC. Complement. Altern. Med. 17, 271-285 (2017) CrossRef PubMed Google Scholar
-
9.Y. Kishimoto, Yakugaku Zasshi 75, 1437-1439 (1955) CrossRef PubMed Google Scholar
-
10.P.R. Diraviam, S.M. Visuvasam, J.B. Alexis, G. Subarayan, U. Toshiyuki, S. Masako, T. Akinobu, F. Hiroyuki, T. Nobutoshi, Chem. Pharm. Bull. 43, 1800-1803 (1995) CrossRef PubMed Google Scholar
-
11.T. Kuraishi, Y. Mitadera, T. Murakami, N. Tanaka, Y. Saiki, C.M. Chen, Yakugaku Zasshi 103, 679-682 (1983) CrossRef PubMed Google Scholar
-
12.K. Yoshiki, M. Masafumi, N. Gen-ichiro, N. Itsuo, Chem. Pharm. Bull. 38, 856-860 (1990) CrossRef PubMed Google Scholar
-
13.A. Tadashi, O. Tadashi, H. Yoshikazu, S. Takayuki, U. Mihoko, O. Shinji, Phytochemistry 46, 839-844 (1997) CrossRef PubMed Google Scholar
-
14.X.L. Li, X. Cheng, L.M. Yang, R.R. Wang, Y.T. Zheng, W.L. Xiao, Y. Zhao, G. Xu, Y. Lu, Y. Chang, Q.T. Zheng, Q.S. zhao, H.D. Sun, Org. Lett. 8, 1937-1940 (2006) CrossRef PubMed Google Scholar
-
15.X.L. Li, L.M. Yang, Y. Zhao, R.R. Wang, G. Xu, Y.T. Zheng, L. Tu, L.Y. Peng, X. Cheng, Q.S. Zhao, J. Nat. Prod. 70, 265-268 (2007) CrossRef PubMed Google Scholar
-
16.X.L. Li, L. Tu, Y. Zhao, L.Y. Peng, G. Xu, X. Cheng, Q.S. Zhao. Helv. Chim. Acta. 91, 856-861 (2008) CrossRef PubMed Google Scholar
-
17.X.Y. Wang, A.N. Gao, Y.D. Jiao, Y. Zhao, X.B. Yang, Int. J. Biol. Macromol. 108, 625-634 (2018) CrossRef PubMed Google Scholar
-
18.A.H. Cory, Cancer Commun. 3, 207-212 (1991) CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2021
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
