


Abietane Diterpernoids from the Roots of Euphorbia ebracteolata
Abstract
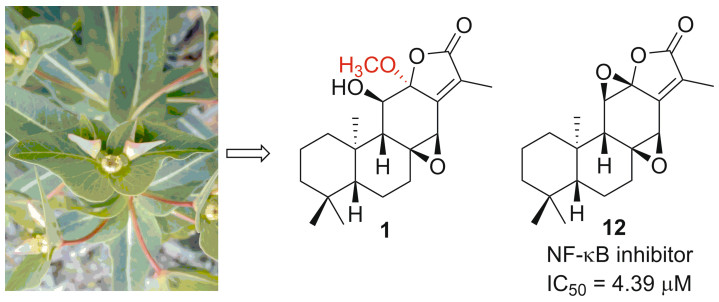
A new ent-abietane diterpernoid, named ebracteolata D (1), along with 11 known analogues, was isolated from the roots of Euphorbia ebracteolata Hayata. The structure of 1 was elucidated on the basis of spectroscopic analysis and molecular modeling. Cytotoxicity of compounds 1-12 was evaluated as well as the effect on the NF-κB pathway. Among them, compound 12, jolkinolide B, displayed broad inhibitory effects against proliferation of tumor cell lines. Mechanistic studies indicated that the compound 12 can inhibit TNF-α induced NF-κB activation, thereby inducing tumor cell apoptosis.Graphical Abstract
Keywords
Euphorbiaceae Euphorbia ebracteolata Abietane diterpernoid Cytotoxic activity1 Introduction
NF-κB is a key transcription factor playing important role in tumor progression and drug resistance [1-3]. Suppression of NF-κB activation could enhance the efficacy of anticancer drugs and overcome drug resistance [4]. Several NF-κB inhibitors have been employed as sensitizers in established cancer therapy over recent years. For example, a combination of NF-κB inhibitor, such as parthenolide or curcumin, with PXL has been demonstrated to augment the therapeutic efficacy in various cancer models [5].
Euphorbiaceae is one of the largest families of higher plants, comprising about 500 genera, 5000 species, and widely distributes in the world, especially in the tropical and subtropical regions [6]. The genus Euphorbia is the largest in the spurge family, some of which have been used as medicinal plants for a long time [7, 8]. Among them, the roots of Euphorbia ebracteolata are used to treat pulmonary tuberculosis, chronic tracheitis, and psoriasis in Traditional Chinese Medicine (TCM) [9]. Till now, a number of diterpenoids with a wide spectrum of bioactivities, including antihepatotoxic and cytotoxic activities, ha ve been isolated from this species [10-12]. In order to identify additional biologically active diterpenoids, a chemical investigation of the roots of E. ebracteolata was performed [13]. As a result, one new ent-abietane diterpernoid, ebracteolata D (1), together with 11 known analogues was obtained from petroleum ether-soluble extracts. The known compounds were identified as 11β-hydroxy-ent-abieta-8(14), 13(15)-dien-16, 12α-olide (2) [14], ebracteolatanolide A (3) [15], yuexiandajisu D (4) [16], yuexiandajisu E (5) [16], ebractenoid K (6) [17], ebractenoid L (7) [17], ebractenoid M (8) [17], tetrahydrojolkinolide B (9) [18], euphorin H (10) [19], methyl-8β, 11β-dihydroxy-12-oxo-ent-abieta-13, 15(17)-dine-16-oate (11) [20] and jolkinolide B (12) [14] by comparison of their spectroscopic data with those reported in the literature. Herein, we report the isolation, structure characterization, and bioactivity evaluation of these compounds (Fig. 1).
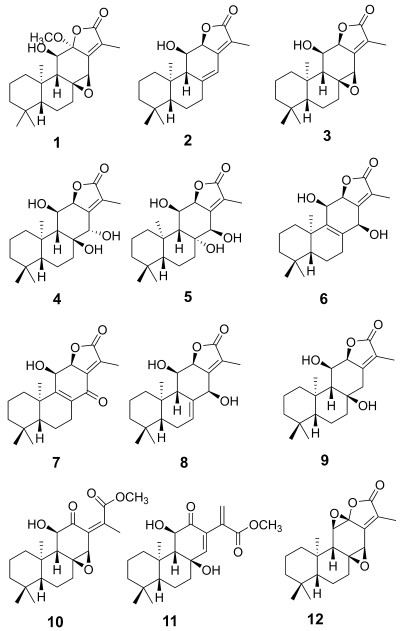
The structures of compounds 1–12
2 Results and Discussion
The 95% EtOH extract of the roots of E. ebracteolata Hayata was fractionated between EtOAc and H2O. The EtOAc fraction was subjected to repeated chromatography, to yield 12 ent-abietane dierpernoids (1–12).
Ebracteolata D (1) was obtained as a whi te powder. The molecular formula was determined as C21H30O5 on the basis of HRESIMS (m/z 385.1993 [M + Na]+), indicating seven degrees of unsaturation. The IR absorptions at 1757 cm−1, in combination with UV absorption maxima at 233, and 308 nm, implied the presence of an α, β-unsaturated-γ-lactone moiety in the molecule, which was confirmed by the 13C NMR signals (δC 169.8, 151.4, and 130.5). The 1D NMR data also afford four methyls, one methoxyl (δH 3.24, s, δc 51.2). five sp3 methylenes, four sp3 oxymethines (δC 73.9/δH 3.69, C-11; δC 57.4/δH 3.77, C-14), and two sp3 oxygenated quaternary carbons (δC 106.5, 65.2). The spectroscopic data and the known chemotaxonomy of this genus suggest a skeleton of ent-abietane diterpernoid (Table 1).
1H (600 MHz) and 13C (150 MHz) NMR spectroscopic data of ebracteolata D (1) in CDCl3
Except for the α, β-unsaturated-γ-lactone moiety, the presence of additional four rings was necessary to meet the required number of unsaturation degrees. Since three rings of an ent-abietane diterpernoid accounted for three degrees of unsaturation, there should be another ring. One oxymethine at δH 3.77 and δC 57.4, along with an oxygenated quaternary carbon at δC 65.2, suggested the presence of an epoxy ring between C-8 and C-14. This was supported by the HMBC correlations from H2-7 (δH 1.58, 2.07) to C-8 and C-14, and from H-14 (δH 3.77) to C-8.
Complete assignments of proton and carbon signals were performed by 2D NMR experiments. According to 1H-1H COSY and HSQC spectrum, the following protonated partial structures were established: C-1/C-2/C-3, C-5/C-6/C-7, and C-9/C-11. The HMBC correlations readily established A and B rings, which were the same as those of jolkinolide B.14 In the HMBC spectrum, cross-peaks of H-11/C-12, and H-14/C-12 and C-13 showed the presence of six-membered ring C, including C-8, 9, 11, 12, 13 and C-14. Moreover, methoxyl at C-12 was determined by the HMBC correlations between OCH3 (δH 3.24, δc 51.2) and C-12. The remaining methyl (δH 8.8, δC 2.03) assigned to the C-17 was linked with C-15 as judged from the correlations from Me-17 to C-13, C-15, and C-16. Although no direct HMBC correlation was available to link C-12 and C-16, from the degrees of unsaturation and their chemical shift, the ester bond between both the carbons could be assumed. The planar structure of Ebracteolata D (1) was thus elucidated as indicated.
The relative configuration of 1 was fixed by ROESY experiment and molecular modeling (Conflex 7 Rev. C). The orientation of Me-18 was arbitrarily assigned as α. In the ROESY spectrum, the cross peaks observed between the proton pairs Me-1/Me-20, H-11/ Me-20 indicated that H-11 and Me-20 were on the same side towards α-orientation (Fig. 2). The ROE correlations of Me-19/H-5, H-5/H-9, and H-9/H-7β suggested that Me-19, H-5, H-9 and H-7β took β-configuration. Moreover, the orientation of the C-8/C-14 epoxy ring was determined to be β due to the ROE correlation between H-7α and H-14, which was identical to that of jolkinolide B. However, due to flexibility of the OCH3 in the structure of 1, it is not sufficient to determine the orientation of the OCH3. Therefore, molecular modeling was performed for the two possible structures of 1, corresponding to the α (A) or β (B) orientation of the OCH3 as shown in Fig. 3. Two optimized structures were obtained, in which the calculated J3-coupling constant between H-9 and H-11 in A was fully consistent with the corresponding NMR data. Therefore, the configuration of OCH3-12 was determined to be α.
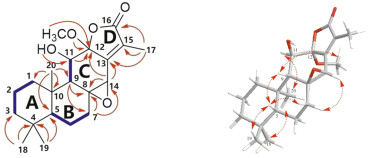
key 1H–1H COSY (


3 Experimental Section
3.1 General Experimental Procedures
Optical rotations data were measured with SEPA-300 and Jasco DIP-370 polarimeter. ESIMS spectra were acquired on Waters 2695 HPLC-Thermo Finnigan LCQ Advantage and HRESIMS API Qstar Pulsar spectrometer. A Bio-Rad FTS-135 spectrophotometer was used for IR spectra as KBr pellets. UV spectra were obtained using a Shimadzu UV-210A spectrophotometer. 1H, 13C NMR and 2D NMR spectra were recorded on Bruker AV-400, DRX-500 and AVANCE Ⅲ-600 MHz spectrometer with TMS as internal standard. Semi-preparative HPLC separations were performed on an Agilent 1100 liquid chromatograph with a Merck i.d. (10 × 100 mm) column. Column chromatography (CC) was performed usi ng silica gel (200–300 mesh and 60–80 mesh, Qingdao Marine Chemical, Inc., Qingdao, P. R. China) and Sephadex LH-20 (40–70 mesh, Amersham Pharmacia Biotech AB, Uppsala, Sweden). Lichroprep RP-18 gel (40–63 μm; Merck, Darmstadt, Germany). And spots were visualized by heating silica gel plates sprayed with 5% H2SO4 in EtOH.
3.2 Plant Material
The roots of E. ebracteolata were collected from Anhui Province, People's Republic of China, in November 2014. The plant samples were identified by Prof. Ji-Ming Xü of Kunming Institute of Botany, Chinese Academy of Sciences (CAS). A voucher specimen (HXJ20141108) was deposited at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences (CAS).
3.3 Extraction and Isolation
The air-dried, powdered plant materials (20 kg) were extracted with 95% EtOH under reflux three times. The combined EtOH extracts were concentrated under vacuum to give a crude residue (2 kg), which was suspended in water and then partitioned with petroleum ether. The petroleum ether portion (780 g) was subjected to passage over a silica gel column, eluted with a gradient of petroleum ether/ethyl acetate (from 100:0 to 0:100), to yield five major fractions (1–5). Fr.2 (60 g) was then chromatographed on a silica gel column eluted with petroleum ether/acetone (25:1), to get 2 (0.6 g), 3 (45 mg) and Fr.2-3. Fr.2-3 was purified by Sephadex LH-20 (Acetone) and semipreparative HPLC to give 1 (5 mg), 10 (11 mg), 11 (4 mg) and 12 (400 mg). Fr3 (65 g) separated over a C18 silica gel column (MeOH/H2O from 2:5 to 4:5) to obtain four further fractions. Fr.3-2 (9 g) was purified by Sephadex LH-20 column (Acetone) to give 6 (20 mg) and 7 (30 mg). Fr3-3 was chromatographed on a silica gel column eluted with petroleum ether/acetone (15:1), and semipreparative HPLC to get 8 (19 mg) and 9 (16 mg). Fr.4 (50 g) was then separated over a C18 silica gel column (MeOH/H2O from 2:5 to 4:5) to obtain four fractions, then Fr.4-1 chromatographed on a silica gel column eluted with petroleum ether/acetone (5:1) and Sephadex LH-20 (Acetone) column to obtain 4 (10 mg) and 5 (28 mg).
3.4 Cytotoxic Activity Assay
The following human tumor cell lines were used: HL-60, SMMC-7721, A-549, MCF-7, and SW-480. All cells were cultured in RPMI-1640 or DMEM medium (Hyclone, Logan, UT, USA), supplemented with 10% fetal bovine serum (Hyclone) at 37℃ in a humidified atmosphere with 5% CO2. Cell viability was assessed by conducting colorimetric measurements of the amount of insoluble formazan formed in living cells based on the reduction of 3-(4, 5-dimethylthiazol-2-yl)-5(3-carboxymethoxyphenyl)-2-(4-sulfopheny)-2H-tetrazolium (MTS) (Sigma, St. Louis, MO, USA). Briefly, 100 mL of adherent cells was seeded into each well of a 96-well cell culture plate and allowed to adhere for 12 h before test compound addition, while suspended cells were seeded just before test compound addition, both with an initial density of 1 × 105 cells/mL in 100 mL of medium. Each cell line was exposed to the test compound at various concentrations in triplicate for 48 h, with cisplatin and paclitaxel (Sigma) used as positive controls. After the incubation, MTS (100 mg) was added to each well, and the incubation continued for 4 h at 37 ℃. The cells were lysed with 100 mL of 20% SDS-50% DMF after removal of 100 mL of medium. The optical density of the lysate was measured at 595 nm in a 96-well microtiter plate reader (Bio-Rad 680). The IC50 values of each compound were calculated by Reed and Muench's method. Activity evaluation methods such as literature method, cisplatin was used as positive control (cisplatin showed cytotoxicity the IC50 values at 18.80 (HL-60), 12.32 (A-549), 17.50 (SMMC-7721), 20.50 (MCF-7), 12.01 (SW-480) μM, respectively).
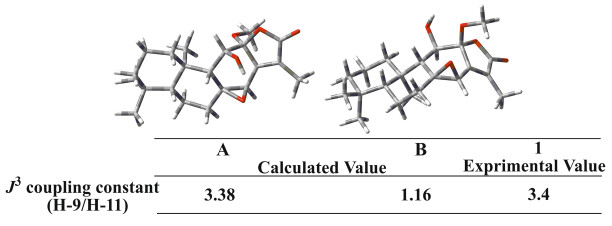
Two possible structures (A and B) of 1. Calculated and experimental J3-coupling constants between H-9 and H-11 are also available
3.5 NF-κB Luciferase Reporter Assay
NF-κB pathway activity was tested by the dual luciferase reporter gene assay method. EK 293T cells were seeded in 24-well plates and transiently transfected with 5 × κB-luciferase and pTK-Renilla reporters using Lipofectamine 2000 for 18 h. Cells were then incubated with different concentrations of compounds for different times, and subsequently stimulated with 10 ng/mL TNF-α for 4 h. Luciferase activity of cell lysate was analyzed using the Dual Luciferase Reporter Assay System (Promega).
Notes
Acknowledgements
This research was supported financially by grants from the National Science Foundation of China (21432010, 81573323, and 31770392), Technological Leading Talent Project of Yunnan Province (2015HA020), Central Asian Drug Discovery and Development Center of Chinese Academy of Sciences (CAM201402, CAM201302).
Compliance with Ethical Standards
Conflict of interest
The authors declare that there is no conflict of interest
References
-
1.M. Karin, Nature 441, 431-436 (2006) CrossRef PubMed Google Scholar
-
2.M.B. Alj, V. Barbu, M. Fillet, A. Chariot, B. Relic, N. Jacobs, J. Gielen, M.P. Merville, V. Bours, Oncogene 22, 90-97 (2003) CrossRef PubMed Google Scholar
-
3.A.S. Baldwin, J. Clin. Invest. 107, 241-246 (2001) CrossRef PubMed Google Scholar
-
4.H.M. Shen, V. Tergaonkar, Apoptosis 14, 348-363 (2009) CrossRef PubMed Google Scholar
-
5.J.M. Barret, A. Bouchou, M.L. Marionneau, V. Offrete, N. Cabrol, C. Bailly, Can. Res. 64, 460-460 (2004) PubMed Google Scholar
-
6.A. Vasas, J. Hohmann, Chem. Rev. 114, 8579-8612 (2014) CrossRef PubMed Google Scholar
-
7.C.S. Huang, S.H. Luo, Y.L. Li, C.H. Li, J. Hua, Y. Liu, S.X. Jing, Y. Wang, M.J. Yang, S.H. Li, Nat. Prod. Bioprospect. 4, 91-92 (2014) CrossRef PubMed Google Scholar
-
8.Y.F. Yang, J.Q. Peng, L. Shi, Z.R. Li, M.H. Qiu, Nat. Prod. Bioprospect. 3, 99-102 (2013) CrossRef PubMed Google Scholar
-
9.Chinese Pharmacopoeia Commission, Pharmacopoeia of the People's Republic of China. I (China Medical Science Press, Beijing, 2010) PubMed Google Scholar
-
10.W.J. Yuan, G.P. Yang, J.H. Zhang, Y. Zhang, D.Z. Chen, S.L. Li, Y.T. Di, X.J. Hao, Phytochem. Lett. 18, 176-179 (2016) CrossRef PubMed Google Scholar
-
11.W.J. Yuan, W.F. Gao, J.H. Zhang, P. Cao, Y. Zhang, D.Z. Chen, S.L. Li, Y.T. Di, X.J. Hao, Nat. Prod. Commun. 12, 63-65 (2017) PubMed Google Scholar
-
12.W. J. Yuan, X. Ding, Z. Wang, B. J. Yang, X. N. Li, Y. Zhang, D. Z. Chen, S. L. Li, Q. Chen, Y. T. Di, H. A. Aisa, X. J. Hao, Sci. Rep. 7, e14507 (2017) PubMed Google Scholar
-
13.Y. Li, M. Xu, X. Ding, C. Yan, Z.Q. Song, L.W. Chen, X.H. Huang, X. Wang, Y.L. Jian, G.H. Tang, C.Y. Tang, Y.T. Di, S.Z. Mu, X.Z. Liu, K. Liu, T. Li, Y.C. Wang, L. Miao, W.X. Guo, X.J. Hao, C.L. Yang, Nat. Cell. Biol. 18, 1065-1077 (2016) CrossRef PubMed Google Scholar
-
14.A.R. Lal, R.C. Cambie, P.S. Rutledge, P.D. Woodgate, Phytochemistry 29, 2239-2246 (1990) CrossRef PubMed Google Scholar
-
15.W.X. Wang, X.B. Ding, Acta. Pharm. Sin. 33, 128-131 (1998) PubMed Google Scholar
-
16.H.M. Shi, L.D. Williams, H.H. Sung, H.X. Zhu, Y.I. Nancy, Z.D. Min, Planta Med. 71, 349-354 (2005) CrossRef PubMed Google Scholar
-
17.Z.G. Liu, Z.L. Li, D.H. Li, N. Li, J. Bai, F. Zhao, D.L. Meng, H.M. Hua, Bioorg. Med. Chem. Lett. 26, 1-5 (2016) CrossRef PubMed Google Scholar
-
18.D. Uemura, Y. Hirata, Tetrahedron Lett. 15, 1387-1390 (1972) PubMed Google Scholar
-
19.X.Z. Kuang, W. Li, Y. Kanno, N. Yamashita, S. Kikkawa, I. Azumaya, K. Nemoto, Y. Asada, K. Koike, J. Nat. Med. 70, 412-422 (2016) CrossRef PubMed Google Scholar
-
20.T. Morgenstern, M. Bittner, M. Silva, P. Aqueveque, J. Jakupovica, Phytochemistry 41, 1149-1153 (1996) CrossRef PubMed Google Scholar
-
21.P. W. Wang, S. M. Henning, D. Herbe, PLoS ONE 5, e10202 (2010) PubMed Google Scholar
-
22.C. Lv, X.H. Yan, Q. Tu, Y.T. Di, C.M. Yuan, X. Fang, Y. BenDavid, L. Xia, J.X. Gong, Y.M. Shen, Z. Yang, X.J. Hao, Angew. Chem. Int. Ed. 55, 7539-7543 (2016) CrossRef PubMed Google Scholar
-
23.Y. Yan, J.X. Zhang, T. Huang, X.Y. Mao, W. Gu, H.P. He, Y.T. Di, S.L. Li, D.Z. Chen, Y. Zhamg, X.J. Hao, J. Nat. Prod. 78, 811-821 (2015) CrossRef PubMed Google Scholar
-
24.X.H. Yan, Y.T. Di, X. Fang, S.Y. Yang, H.P. He, S.L. Li, Y. Lu, X.J. Hao, Phytochemistry 72, 508-513 (2011) CrossRef PubMed Google Scholar
-
25.F.G. Zeng, Q. Su, Y.T. Di, X.J. Hao, Nat. Prod. Res. Dev. 28, 1171-1175 (2016) PubMed Google Scholar
Copyright information
© The Author(s) 2018
Open Access
This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creative commons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
