


Total synthesis and determination of the absolute configuration of a natural analgesic: crotonine
Abstract
We report the first total synthesis of four possible absolute configurations and four other regional isomers of a naturally occurring alkaloid crotonine, which was isolated from Croton tiglium L. (Euphoriaceae) without elucidation of its absolute configuration. The concise five-step route with a chirally poor and regioselective strategy starting from monosaccharides was established, and the absolute structure of the natural crotonine was determined by comparison of the NMR spectra and optical rotations of the synthetic products.Keywords
total synthesis crotonine 2-(furan-2-yl)-5-(2, 3, 4-trihydroxy-butyl)-1, 4-diazine Croton tiglium L. (Euphoriaceae) analgesicsIntroduction
Pyrazines are important components of processed foods and are associated with the generation of flavor and color, such as those of fermented cacao, cheese and wines.1 The Maillard reaction of amino acids with reducing sugars has become the primary process for generating these pyrazine derivatives in food processing.2 Some molecules containing the pyrazine pharmacophore have exhibited remarkable pharmacological activities with antitumor, 3 diuretic, 4 immunomodulatory5 and analgesic effects.6 Crotonine (2-(furan-2-yl)-5-(2, 3, 4-trihydroxy-butyl)-1, 4-diazine) was isolated from Crotontiglium L. (Euphoriaceae) without determination of the absolute configuration. However, the analgesic properties were obvious compared to those of morphine.7 To determine the absolute structure of natural crotonine, a concise saccharide-based fivestep route was developed for the synthesis of all possible chiral isomers. The synthesis of pyrazine derivatives has attracted much interest due to their numerous scientific applications in both chemistry and biology. The condensation reaction of diamines with diols, 8 hydroxyketones9 and diketones10 under catalysis by a transitional metal or base are the main reaction types reported in the literature. In our case, inexpensive monosaccharides were adopted as the starting material and chirality source for the preparation of α-dicarbonyl intermediates.11 Pyrazine condensation of the diamine and 3-deoxy-aldos-2-uloses intermediates, which is the key step in the reaction sequence, was catalyzed by potassium hydroxide, and other reaction conditions were successively investigated. The retrosynthetic analysis is outlined in Scheme 1.
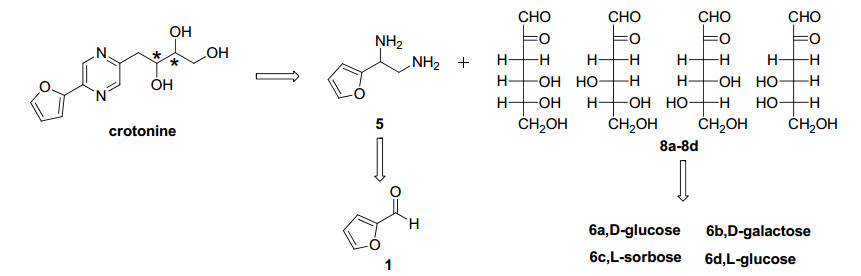
Retrosynthetic analysis of crotonine
Results and Discussion
The preparation of the 1, 2-ethylenediamine intermediate 5 was initiated with furfuraldehyde 1 (Scheme 2).12 Henry addition with nitromethane generated 2-(2-nitroethenyl)furan 2 in 72% yield, which was followed by treatment with methoxylamine hydrochloride to yield N-methoxyethylamine 3 in 88% yield. According to the published procedure, reduction of 3 by zinc powder and acetic acid was attempted to generate 1, 2-ethylenediamine 5 directly, however, this reaction only yielded a series of partially reduced byproducts and was difficult for purification.12 While catalytic hydrogenation over Pd/C only afforded partially reduced monoamine at the nitro group, when the Pd/C-catalyzed reaction was conducted under higher pressure or temperature, or a more active reducing reagent such as palladium hydroxide was used, the reduction of 3 produced an over-reduced product at the furan moiety. An exchange strategy succeeded in cleaving the methoxyl group from the nitrogen to yield nitro-ethenamine 4 in 95% yield, 13 followed by reduction with LiAlH4 in THF to afford 1, 2-ethylenediamine intermediate 5 in 52% yield.
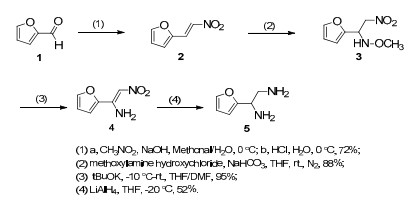
Synthesis of diamine intermediate 5
Construction of the 3-deoxy-aldos-2-ulose intermediates 8a–8d started with monosaccharides (Scheme 3), such as Dglucose 6a, D-galactose 6b, L-sorbose 6c and L-glucose 6d, which possess different potential chiral centers at the 2" and 3" positions in the final product. Refer to the method described in the literature, 11 the monosaccharides reacted with benzoyl hydrazine to yield 3-deoxy-bis(benzoylhydrazones) 7a–7d in 63%–93% yield. The mechanism was explained as a repeated intramolecular rearrangement and dehydration process of the 1 and 2 position of the monosaccharide.11 After easily obtaining the products 7a–7d by filtration, a transhydrazonation reaction with benzaldehyde afforded 3-deoxy-aldos-2-uloses 8a–8d in 25%–37% yield.
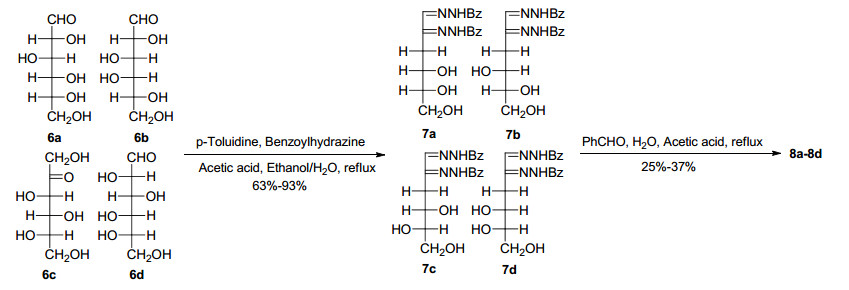
Synthesis of intermediates 8a–8d
The condensation of the two key intermediates (i.e., 5 and 8a–8d) was exhibited in Scheme 4, the reaction of 5 and 8 without base conducted under an argon atmosphere was unsuccessful, 14 because the alkalinity of 5 was insufficient to spontaneously catalyze the cyclization. When the strong base as potassium tert-butoxide or potassium hydroxide was used under an argon and anhydrous conditions, 15 a trace product was obtained, but the yield failed to increase even with a high temperature and long reaction time. Next, the reaction was attempted in an air atmosphere using potassium hydroxide at 0 ºC, but no reaction occurred.16 Fortunately, the yield of 9 was improved after increasing the temperature to room temperature, but the regional isomers 10 were also produced, the quantity of product 9 and 10 were in a 1:1 ratio (the ratio was determined by 1H NMR analysis). After we screened the reaction conditions, including varying the quantity of the reagents and the addition method, it was determined that the addition frequency of potassium hydroxide markedly influenced the regioselectivity of the pyrazine condensation. In the synthesis of 9 and 10, room temperature and multiple additions of potassium hydroxide (i.e., 10 portions of solid for 1 hour) favored the generation of a higher ratio of para-substitution 9 to meta-substitution 10 products (i.e., up to 5:1), and finally all 8 regional and chiral isomers were synthesized and purified successively.
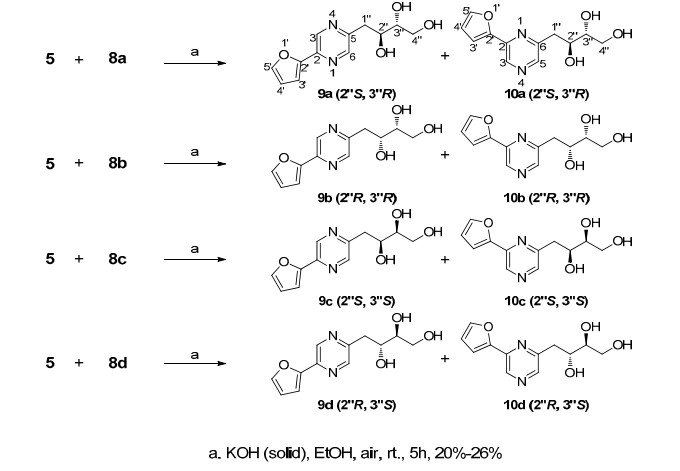
Synthesis of 9 and 10
A comparison of the spectroscopic data of the natural7 and synthetic products was conducted (Tables 1 and 2). The 1H NMR of 10a–10d differed from those of the natural product which exhibited a singlet peak for H-3 and H-5 of the pyrazine ring, indicating that 10a–10d were meta-substitution products; while 9a–9d and natural crotonine showed a doublet peak (J = 1.1 to 1.4 Hz) which attributed them to the para-substitution products.17 The remaining 1H and 13C NMR data of 10a–10d were similar to that of 9a–9d, inferring that each pair of 9 and 10 (i.e., 9a and 10a) were regional isomers and possessed the same chirality at 2" and 3" position which were controlled by the chirality of intermediates 8a–8d. To the para-substitution products 9a–9d, the characteristic proton signal of 1" of 9b and 9c crotonine showed only one set of doublet peak, that differed obviously from that of 9a, 9d and the natural crotonine which exhibited two set of dd peaks, in addition, the 1H and 13C NMR data for 9a and 9d matched those of the reported natural product exactly, indicating that the absolute configuration of natural crotonine only could be equal to 9a or 9d. A further comparison of the optical rotation of 9a and 9d indicated that that of 9a (2"S, 3"R)-crotonine ([α]D24 – 30.0 (c 0.17, MeOH)) was closer to that of the natural product ([α]D20 – 6.8 (c 0.25, MeOH)), because the enantiomer 9d (2"R, 3"S)-crotonine ([α]D25 + 23.5 (c 0.18, MeOH)) produced an opposite optical rotation that was substantially different from that of the natural product. Therefore, we inferred that the most likely absolute configuration of crotonine was identical to 9a as 2-(furan-2'-yl)-5-(2"S, 3"R, 4"-trihydroxy-butyl)-1, 4-diazine.
1H NMR data of natural crotonine and synthetic products in CD3OD
13C NMR data of crotonine and synthetic products in CD3OD
In conclusion, we devised a concise five-step saccharidebased procedure to synthesize all eight possible regional and chiral isomers of crotonine for the first time. This method may be conveniently extended to other diamines and monosaccharides for the synthesis of a series of potentially bioactive pyrazines. Then, we determined that the most probable absolute configurations of the natural product by comparison of the NMR spectra and optical rotation data.
Experimental Section
General Experimental Procedures. All reactions were performed with chemically pure solvents without further purifications unless otherwise noted. Dry solvent was obtained by a standard procedure. Dry tetrahydrofuran (THF) was distilled over a sodium-potassium alloy. Diethylformamide (DMF) was distilled over calcium hydride. Yields refer to chromatographically and spectroscopically homogeneous materials, unless otherwise noted. Reagents were used as received without further purification. Silica gel (200–300 mesh, Qingdao Marine Chemical Ltd., China), and light petroleum ether (bp 60–90 ºC), ethyl acetate and methanol were used for product purification used for flash column chromatography. Purity of compounds were determined by Agilent 1200 Series HPLC using Thermo Hypersil Gold C18 column eluted with a mixture of methanol and water. NMR spectra were recorded in CDCl3, C5D5N and Methanol solutions on Bruker AV-400/800 and Bruker DRX-500 instrument with tetramethylsilane (TMS) as an internal reference. IR spectra were recorded with KBr pellets on a Bruker Tensor 27 FT-IR spectrometer. UV data were obtained on a Shimadzu UV-2401A spectrophotometer. Highresolution mass spectral analysis (HRMS) data were recorded via electron impact mass spectrometry using a time of flight analyzer. Optical rotations were determined on a Jasco P-1020 digital polarimeter.
2-(2-Nitroethenyl)furan (2). According to the literature procedure, 12 to a 2 L three neck flask was added 1 (100 g, 1.04 mol) and nitromethane (63.5 g, 1 equivalent) before 200 mL methanol was added at 0 ºC, an aqueous sodium hydroxide (44 g, 1.1 equivalent) dissolved in 200 mL water was dropped into the mixture slowly to maintain the system stirring below 0 ºC, then maintain the mixture stirring for 1 hour at 0 ºC. Then 200 mL ice-water was poured into the mixture to generate a brown solution. To a cold solution prepared by 300 mL concentrated hydrochloride and 300 mL water, the former brown solution was added in slowly to furnish a yellow solid. Filtrated and washed by water several times, afforded the crude product, after repeated crystallization with methanol affording 2 as a yellow needle crystal 103 g (72%). 1H NMR (400 MHz, CDCl3)δ ppm: 7.74 (d, J = 13.2 Hz, 1H), 7.56 (dd, J = 1.2, 0.5 Hz, 1H), 7.47 (d, J = 13.2 Hz, 1H), 6.88 (d, J = 3.5 Hz, 1H), 6.55 (dd, J = 3.5, 1.8 Hz, 1H).
1-(Furan-2-yl)-2-nitro-N-methoxyethylamine (3). To a mixture of 2 (102 g, 0.73 mol), methoxylamine hydroxychloride (74 g, 1.2 equivalent) and sodium bicarbonate (74 g, 1.2 equivalent) was dissolved with 150 mL water and stirred, at the end of carbon dioxide generation, 650 mL THF was added and discharged the atmosphere with nitrogen for three times, stirred at room temperature overnight. When the reaction was complete, seperated 3 times by 200 mL ethyl acetate before combined the organic layer and concentrated the solvent, filtrated and affordded a yellow liquid 92 g (88%). 1H NMR (400 MHz, CDCl3) δ ppm: 7.37 (m, 1H), 6.34 (m, 1H), 6.31 (d, J = 2.5 Hz, 1H), 5.74 (br. s, 1H), 4.96–4.75 (m, 2H), 4.69 (dd, J = 11.9, 4.2 Hz, 1H), 3.46 (s, 3H); 13C NMR (100 MHz, CDCl3) δ ppm: 148.9, 142.6, 110.4, 108.4, 74.5, 62.4, 56.3.
1-(Furan-2-yl)-2-nitro-ethenamine (4). Added a solution of 3 in 50 mL dry DMF (22 g, 118.3 mmol) slowly to t-BuOK (29.2 g, 2.2 equivalent) dissolved in 200 mL anhydrous THF, maitaning the temperature at – 10ºC, then keep stirring for 5 h at room temprature, seperated by ethyl acetate and water for 5 times, combined the organic layer and concentrated the solvent after dried over with sodium sulfate, and the crude product was purified by recrystallization with methanol, dried under air to generate 4 as a brown solid 17.3 g (95%). 1H NMR (400 MHz, CDCl3) δ ppm: 7.64 (d, J = 1.1 Hz, 1H), 7.08 (s, 1H), 6.96 (d, J = 3.6 Hz, 1H), 6.59 (dd, J = 3.6, 1.7 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ ppm: 145.9, 145.1, 113.0, 112.8, 108.4; ESIMS: 155 [M + H]+, 177 [M + Na]+.
1-(Furan-2-yl)-1, 2-ethylenediamine (5). To a solution of LiAlH4 in 200 mL dry THF was dropped in a solution of 4 in 50 mL THF at a liquid nitrogen-acetone bath maintaining the temprature below – 10 ºC, the reaction finished after 5 hours at 0 ºC, quenched with water very carefully, filtrated and affored a yellow solution, purified by RP-18 column and gave 5 as a brown liquid 11.2 g (90 mmol, 52%), conserved at – 20 ºC under nitrogen. 1H NMR (400 MHz, CDCl3) δ ppm: 7.35 (m, 1H), 6.28 (m, 1H), 6.17 (m, 1H), 3.95 (m, 1H), 3.04 (dd, J = 12.5, 4.2 Hz, 1H), 2.93 (dd, J = 7.7, 3.8 Hz, 1H), 2.14 (br. s, 4H); 13C NMR (100 MHz, CDCl3): δ ppm: 157.0, 141.7, 110.1, 105.1, 51.7, 46.7; HRMSEI m/z: calcd for C6H10N2O [M]+: 126.0793, found: 126.0796.
3-Deoxy-D-erythro-hexos-2-ulose bis(benzoylhydrazone) (7a). Followed the methods of Khadem, 11e to a solution of 600 mL ethanol and 120 mL water was added D-glucose 6a (30 g, 0.17 mmol), benzoylhydrazine (46 g, 0.34 mmol) and ptoluidine (12 g, 0.11 mmol) followed by 50 mL acetic acid, refluxed for 7 hours. Filtrated afforded a white solid, washed by water and ethanol, then a crystallization with ethanol generated 7a as a white needle 44.6 g (0.12 mmol, 71%). [α]D24 + 4.8 (c 0.56, Pyridine); 1H NMR (400 MHz, C5D5N) δ ppm: 14.61 (s, 1H), 14.33 (s, 1H), 10.41 (s, 1H), 9.71 (m, 4H), 9.00–8.82 (m, 6H), 6.24 (br. s, 1H), 5.98–5.66 (m, 3H), 5.14 (br. s, 2H); 13C NMR (100 MHz, C5D5N) δ ppm: 167.3, 166.5, 158.2, 151.1, 136.1, 135.8, 134.3, 131.0, 130.8, 130.5, 130.2, 77.0, 75.1, 66.3, 32.6; HRMSEI m/z: calcd for C20H22N4O5 [M]+: 398.1590, found 398.1568.
3-Deoxy-D-threo-hexos-2-ulose bis(benzoylhydrazone) (7b). The procedure was conducted as the synthsis of 7a, D-galactose 6b 10 g, benzoylhydrazine 15.4 and p-toluidine 4 g, generated 7b as a white needle 15.57 g (80%). [α]D24 + 47.0 (c 0.42, Pyridine); 1H NMR (400 MHz, C5D5N) δ ppm: 14.48 (s, 1H), 14.35 (s, 1H), 10.30 (s, 1H), 9.81 (br. d, J = 7.1 Hz, 2H), 9.69 (br. d, J = 7.7 Hz, 2H), 8.89 (m, 6H), 6.33 (br. s, 1H), 5.96–5.66 (m, 3H), 5.31–5.01 (m, 2H); 13C NMR (100 MHz, C5D5N) δ ppm: 167.1, 166.1, 157.6, 150.7, 136.3, 134.1, 130.9, 130.7, 130.4, 130.2, 77.2, 73.6, 66.4, 33.4; HRMSEI m/z: calcd for C20H22N4O5 [M]+: 398.1590, found 398.1601.
3-Deoxy-L-threo-hexos-2-ulose bis(benzoylhydrazone) (7c). The procedure was conducted as the synthsis of 7a, L-sorbose 6c 10 g, benzoylhydrazine 15.4 g and p-toluidine 4 g, generated 7c as a white needle 18.5 g (93%). [α]D24 – 51.2 (c 0.54, Pyridine); 1H NMR (400 MHz, C5D5N)δ ppm: 14.48 (s, 1H), 14.35 (s, 1H), 10.31 (s, 1H), 9.81 (br. d, J = 7.1 Hz, 2H), 9.69 (br. d, J = 7.7 Hz, 2H), 8.89 (m, 6H), 6.33 (br. s, 1H), 5.87 (m, 2H), 5.72 (m, 1H), 5.36–5.09 (m, 2H); 13C NMR (100 MHz, C5D5N) δ ppm: 167.1, 166.3, 157.6, 150.7, 136.3, 134.1, 130.9, 130.8, 130.4, 130.2, 77.2, 73.6, 66.4, 33.4; HRMSEI m/z: calcd for C20H22N4O5 [M]+: 398.1590, found 398.1592.
3-Deoxy-L-erythro-hexos-2-ulose bis(benzoylhydrazone) (7d). The procedure was conducted as the synthsis of 7a, Lglucose 6d 1 g, benzoylhydrazine 1.54 g and p-toluidine 0.4 g, generated 7d as a white needle 1.25 g (63%). [α]D24 – 3.4 (c 0.45, Pyridine); 1H NMR (400 MHz, C5D5N) δ ppm: 14.60 (s, 1H), 14.27 (s, 1H), 10.27 (s, 1H), 9.70 (br. d, J = 7.1 Hz, 2H), 9.62 (br. d, J = 7.4 Hz, 2H), 8.92–8.84 (m, 2H), 8.80 (m, 4H), 6.20 (br. s, 1H), 5.93–5.61 (m, 3H), 5.07 (br. s, 2H); 13C NMR (100 MHz, C5D5N) δ ppm: 167.2, 166.2, 157.8, 151.0, 136.1, 135.7, 134.3, 130.9, 130.7, 130.3, 130.0, 76.8, 75.0, 66.2, 32.4; HRMSEI m/z: calcd for C20H22N4O5 [M]+: 398.1590, found 398.1588.
3-Deoxy-D-erythro-hexos-2-ulose (8a). Followed the methods of Khadem, 11d to 300 mL methanol was added 7a (20 g, 54 mmol) and 500 mL water, after acetic acid (12 mL) and benzaldehyde (32 mL) were added in, the mixture was to refluxed for 2 hours, when the hydrazone was completely dissovled, the reaction was contiued to refluxed for another 10 hours with vigrously. Filtrated and the solution was concentrated in vaccour, the residue was seperated with water and dichloromethane by 5 times, combined the aqueous part and evaperated the solvent, furnished 8a as a yellow colloidal solid 3.2 g (32 mmol, 37%); 1H NMR (400 MHz, CD3OD) δ 4.42 (br. s, 1H), 4.39 (br. s, 1H), 4.37 (br. s, 1H), 4.30 (br. s, 1H), 4.28 (br. s, 1H), 4.24 (d, J = 2.7 Hz, 1H), 4.18 (m, 1H), 4.05 (m, 1H), 4.00 (m, 1H), 3.97 (m, 1H), 3.90 (d, J = 2.8 Hz, 1H), 3.86 (m, 1H), 3.80 (m, 1H), 3.69 (m, 4H), 3.65 (m, 2H), 3.61 (m, 2H), 3.57 (m, 1H), 3.53 (m, 1H), 3.42 (dd, J = 3.9, 1.7 Hz, 2H), 3.30 (dd, J = 5.5, 3.9 Hz, 1H), 2.45 (m, 1H), 2.16 (m, 3H), 1.99 (m, 1H), 1.85 (m, 2H), 1.76 (m, 2H); 13C NMR (100 MHz, CD3OD) δ 172.4, 172.0, 170.9, 169.7, 106.8, 106.7, 106.5, 106.2, 100.7, 100.1, 100.0, 99.9, 98.4, 98.3, 93.7, 93.3, 88.4, 88.3, 87.3, 87.2, 74.3, 74.0, 73.7, 73.6, 72.5, 72.2, 72.1, 72.0, 69.2, 68.8, 67.8, 66.6, 65.3, 65.2, 65.1, 65.0, 63.9, 63.8, 63.2, 62.9, 62.8, 62.8, 60.3, 60.3, 55.7, 55.7, 55.7, 55.6, 42.89, 42.4, 42.1, 41.9, 38.6, 34.2, 33.0, 32.6. [α]D24 + 11.0 (c 0.38, H2O); HRMSEI m/z: calcd for C6H10O5 [M]+: 162.0528, found: 162.0534.
3-Deoxy-D-threo-hexos-2-ulose (8b). The procedure was conducted as the synthsis of 8a. 7b 15.57 g, acetic acid (18.7 mL) and benzaldehyde (25 mL), afforded a yellow colloidal solid 1.96 g (25%); 1H NMR (400 MHz, CD3OD) δ 4.44 (m, 1H), 4.41 (br. s, 0.5H), 4.39 (br. s, 0.5H), 4.34 (m, 0.5H), 4.32 (br. s, 1H), 4.30 (br. s, 1H), 4.26 (m, 0.5H), 4.19 (m, 1.5H), 4.07 (m, 1.5H), 4.02 (d, J = 5.0 Hz, 1.5H), 4.00 (d, J = 1.2 Hz, 1.5H), 3.88 (dd, J = 4.5, 2.8 Hz, 1H), 3.83 (d, J = 2.3 Hz, 0.5H), 3.80 (m, 0.5H), 3.72 (m, 5H), 3.67 (m, 2.5H), 3.62 (m, 1.5H), 3.59 (d, J = 5.0 Hz, 1.5H), 2.46 (dt, J = 13.7, 7.5 Hz, 1H), 2.17 (m, 3H), 2.02 (m, 0.6H), 1.82 (m, 4H); 13C NMR (150 MHz, CD3OD) δ 172.4, 172.2, 171.7, 171.3, 105.5, 105.4, 105.2, 105.0, 99.5, 99.4, 98.8, 98.8, 98.7, 87.2, 86.0, 85.9, 71.3, 71.0, 70.8, 70.7, 68.0, 65.4, 63.9, 63.9, 62.6, 62.6, 61.9, 61.5, 41.6, 41.2, 40.9, 40.7, 37.4, 33.0, 31.8, 31.3; [α]D24 – 19.8 (c 0.41, H2O); HRMSEI m/z: calcd for C6H10O5 [M]+: 162.0528, found: 162.0531.
3-Deoxy-L-threo-hexos-2-ulose (8c). The procedure was conducted as the synthsis of 8a. 7c 8.54 g, acetic acid 10.3 mL and benzaldehyde 13.7 mL, afforded 8c as a yellow colloidal solid 1.07 g (31%); 1H NMR (400 MHz, CD3OD) δ 4.44 (br. s, 1H), 4.41 (br. s, 0.5H), 4.39 (s, 0.5H), 4.30 (m, 1H), 4.32 (d, J = 4.5 Hz, 1H), 4.29 (d, J = 4.5 Hz, 1H), 4.27 (d, J = 2.6 Hz, 1H), 4.19 (m, 0.5H), 4.06 (m, 1H), 4.02 (d, J = 5.3 Hz, 1H), 4.00 (m, 1H), 3.88 (m, 1H), 3.83 (dd, J = 5.0, 2.7 Hz, 1H), 3.80 (m, 1.5H), 3.70 (m, 3.5H), 3.63 (m, 2H), 3.56 (m, 2.5H), 2.46 (m, 1H), 2.15 (m, 2H), 2.02 (m, 0.5H), 1.81 (m, 4H); 13C NMR (100MHz, CD3OD) δ 170.1, 170.0, 169.6, 169.3, 105.5, 105.4, 105.2, 105.0, 99.5, 99.5, 98.8, 98.8, 98.7, 87.2, 87.0, 86.0, 85.9, 71.3, 71.0, 70.8, 67.9, 65.4, 65.3, 64.0, 63.9, 62.6, 62.6, 61.9, 61.6, 41.6, 41.2, 40.9, 40.7, 35.2, 33.0, 31.8, 31.4; [α]D24 + 12.2 (c 0.55, H2O); HRMSEI m/z: calcd for C6H10O5 [M]+: 162.0528, found: 162.0537.
3-Deoxy-L-erythro-hexos-2-ulose (8d). The procedure was conducted as the synthsis of 8a. 7d 1.15 g, acetic acid 1.38 mL and benzaldehyde 1.85 mL, afforded 8d as a yellow colloidal solid 104 mg (23%); 1H NMR (400 MHz, CD3OD) δ 4.44 (m, 1H), 4.41 (d, J = 3.1 Hz, 0.5H), 4.39 (m, 0.5H), 4.32 (br. s, 1H), 4.30 (br. s, 1H), 4.20 (m, 1H), 4.08 (dt, J = 9.4, 4.2 Hz, 1.5H), 4.02 (dd, J = 6.5, 3.7 Hz, 2H), 4.00 (d, J = 4.5 Hz, 2H), 3.89 (m, 1H), 3.72 (m, 5H), 3.68 (m, 3H), 3.62 (m, 3H), 2.45 (m, 1H), 2.18 (m, 2.5H), 1.84 (m, 5H); 13C NMR (100 MHz, CD3OD) δ 171.2, 170.4, 170.2, 169.8, 105.6, 105.4, 105.3, 105.0, 99.5, 99.5, 98.9, 98.8, 98.7, 87.1, 87.0, 86.0, 85.9, 71.3, 70.9, 71.0, 67.9, 65.4, 65.4, 63.9, 62.7, 62.6, 61.6 54.5, 54.5, 54.4, 41.6, 41.2, 40.9, 40.7, 33.1, 31.8, 31.4; [α]D24 – 10.2 (c 0.62, H2O); HRMSEI m/z: calcd for C6H10O5 [M]+: 162.0528, found: 162.0529.
Synthesis of 9 and 10. To 1.5 equivalent 8 dissolved in ethaol was added 1.0 equivalent 5 in ethanol, allowed the mixture to stirred for 30 miniuts before pattasium hydroxide (3.0 equivalent) was added in 10 portions for 1 hour at room temperature, and kept reaction for a further 5 hours. Quanched by saturated ammonium chloride aqueous solution and evaporated the ethanol, after 20 mL water was added, extracted the low polarity impurities with dichloromethane for one time, the aquous phase was extrated with n-bultyl alchohol for 5 times, combined the organic phase and concentrated under vaccuum, seperated the product by silica gel column and obtained the mixed 9 and 10 isomers, after seperating by repeated Preparative Thin-Layer Chromatography with an eluent (ethyl acetate/methanol/aqueous ammonia = 98:2:5) and affordded the final sturcture 9 and 10 successively.
Synthesis of 9a and 10a. 500 mg 5, 964 mg 8a and 666 mg pattasium hydroxide followed the general procedure A and afforded 261 mg yellow solid (26%), took out 60 mg and seperated by PTLC, obtained 9a (11.5 mg) and 10a (14 mg) as yellow solid successively.
2-(Furan-2'-yl)-5-(2"S, 3"R, 4"-trihydroxy-butyl)-1, 4-diazine (9a). 1H and 13C NMR see Tables 1 and 2; [α]D24 – 30.0 (c 0.17, MeOH); UV (MeOH) λmax(log ε): 272 (4.17) nm; IR (KBr) νmax cm–1: 3424, 2925, 1630, 1498, 1033, 884, 745, 591; 1H and 13C NMR data, see Tables 1 and 2; HREIMS m/z: calcd for C12H14N2O4[M]+: 250.0954, found: 250.0951.
2-(Furan-2'-yl)-6-(2"S, 3"R, 4"-trihydroxy-butyl)-1, 4-diazine (10a). 1H and 13C NMR see Tables 1 and 2; [α]D25 – 60.3 (c 0.09, MeOH); UV (MeOH) λmax(log ε): 328 (4.15) nm; IR (KBr) νmax cm–1: 3406, 2923, 2884, 1630, 1608, 1531, 1491, 1154, 1068, 884, 749, 593; 1H and 13C NMR data, see Tables 1 and 2; HREIMS m/z: calcd for C12H14N2O4 [M]+: 250.0954, found: 250.0961.
Synthesis of 9b and 10b. 50 mg 5, 95 mg 8b and 66 mg pattasium hydroxide followed the general procedure and afforded 23 mg yellow solid (25%), seperated by PTLC and obtained 9b (3.3 mg) and 10b (10 mg) as yellow solid successively.
2-(Furan-2'-yl)-5-(2"R, 3"R, 4"-trihydroxy-butyl)-1, 4-diazine (9b). 1H and 13C NMR see Table 1 and Table 2; [α]D24 + 58.7 (c 0.07, MeOH); UV (MeOH) λmax(log ε): 272 (4.14) nm; IR (KBr) νmax cm–1: 3421, 2925, 1631, 1501, 1384, 1113, 1077, 1037, 746, 593; 1H and 13C NMR data, see Tables 1 and 2; HREIMS m/z: calcd for C12H14N2O4 [M]+: 250.0954, found: 250.0964.
2-(Furan-2'-yl)-6-(2"R, 3"R, 4"-trihydroxy-butyl)-1, 4-diazine (10b). 1H and 13C NMR see Tables 1 and 2; [α]D25 + 44.1 (c 0.15, MeOH); UV (MeOH) λmax(log ε): 328 (4.70) nm; IR (KBr) νmax cm–1: 3421, 2956, 2927, 1630, 1605, 1529, 1491, 1408, 1153, 1091, 1039, 1018, 752; 1H and 13C NMR data, see Tables 1 and 2; HREIMS m/z: calcd for C12H14N2O4 [M]+: 250.0954, found: 250.0950.
Synthesis of 9c and 10c. 50 mg 5, 95 mg 8c and 66 mg pattasium hydroxide followed the general procedure and afforded a 19 mg yellow solid (20%), seperated by PTLC and obtained 9c (1.5 mg) and 10c (6.4 mg) as yellow solid successively.
2-(Furan-2'-yl)-5-(2"S, 3"S, 4"-trihydroxy-butyl)-1, 4-diazine (9c). 1H and 13C NMR see Tables 1 and 2; [α]D25 – 50.4 (c 0.18, MeOH); UV (MeOH) λmax(log ε): 273 (4.03) nm; IR (KBr) νmax cm–1: 3423, 2928, 1630, 1604, 1501, 1111, 1059, 1033, 751, 593; 1H and 13C NMR data, see Tables 1 and 2; HREIMS m/z: calcd for C12H14N2O4 [M]+: 250.0954, found: 250.0958.
2-(Furan-2'-yl)-6-(2"S, 3"S, 4"-trihydroxy-butyl)-1, 4-diazine (10c). 1H and 13C NMR see Tables 1 and 2; [α]D24 – 36.9 (c 0.12, MeOH); UV (MeOH) λmax(log ε): 328 (3.96) nm IR (KBr) νmax cm–1: 3423, 2927, 1630, 1529, 1491, 1153, 1091, 1039, 1018, 752, 595; 1H and 13C NMR data, see Tables 1 and 2; HREIMS m/z: calcd for C12H14N2O4 [M]+: 250.0954, found: 250.0958.
Synthesis of 9d and 10d. 30 mg 5, 58 mg 8d and 40 mg pattasium hydroxide followed the general procedure and afforded 13 mg yellow solid (22%), seperated by PTLC and obtained 9d (1.5 mg) and 10d (1.8 mg) as yellow solid successively.
2-(Furan-2'-yl)-5-(2"R, 3"S, 4"-trihydroxy-butyl)-1, 4-diazine (9d). 1H and 13C NMR see Tables 1 and 2; [α]D25 + 23.5 (c 0.18, MeOH); UV (MeOH) λmax(log ε): 272 (4.27) nm; IR (KBr) νmax cm–1: 3423, 2926, 1630, 1498, 1135, 1109, 1033, 746, 591; 1H and 13C NMR data, see Tables 1 and 2; HREIMS m/z: calcd for C12H14N2O4 [M]+: 250.0954, found: 250.0948.
2-(Furan-2'-yl)-6-(2"R, 3"S, 4"-trihydroxy-butyl)-1, 4-diazine (10d). 1H and 13C NMR see Tables 1 and 2; [α]D25 + 65.3 (c 0.24, MeOH); UV (MeOH) λmax(log ε): 328 (3.95) nm; IR (KBr) νmax cm–1: 3417, 2922, 2883, 1631, 1531, 1491, 1409, 1154, 1068, 1038, 1015, 884, 749, 593; 1H and 13C NMR data, see Tables 1 and 2; HREIMS m/z: calcd for C12H14N2O4 [M]+: 250.0954, found: 250.0961.
Notes
Electronic Supplementary Material
Supplementary material is available in the online version of this article at http://dx.doi.org/10.1007/s13659-013-0080-1and is accessible for authorized users.
Acknowledgments
This work was supported financially and inspired scientifically by Prof. Yongxian Cheng of State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, and Prof. Baomin Fan of Key Laboratory of Chemistry in Ethnic Medicinal Resources, State Ethnic Affairs Commission & Ministry of Education, Yunnan University of Nationalities.
References
-
1.(a) Mihara, S. ; Masuda, H. J. Agric. Food Chem. 1988, 36(6), 1242-1247; (b) Gill, M. S. ; Macleod, A. J. ; Moreau, M. Phytochemistry 1984, 23(9), 1937-1942. PubMed Google Scholar
-
2.(a) Hwang, H. I. ; Hartman, T. G. ; Rosen, R. T. ; Ho, C. T. J. Agric. Food Chem. 1993, 41(11), 2112-2115; (b) Martin, F. L. ; Ames, J. M. J. Agric. Food Chem. 2001, 49(8), 3885-3892. PubMed Google Scholar
-
3.S. Urban, S. J. H. Hickford, J. W. Blunt, M. H. G. Munro, Curr. Org. Chem. 4(7), 765-807 (2000) CrossRef PubMed Google Scholar
-
4.E. J.Jr. Cragoe, O. W.Jr. Woltersdorf, J. B. Bicking, S. F. Kwong, J. H. Jones, J. Med. Chem. 10(1), 66-75 (1967) CrossRef PubMed Google Scholar
-
5.A. Zhu, J. B. Huang, A. Clark, R. Romero, H. R. Petty, Carbohydr. Res. 342(18), 2745-2749 (2007) CrossRef PubMed Google Scholar
-
6.Y. Jinsmaa, Y. Okada, Y. Tsuda, K. Shiotani, Y. Sasaki, A. Ambo, S. D. Bryant, L. H. Lazarus, J. Pharmacol. Exp. Therap. 309(1), 432-438 (2004) CrossRef PubMed Google Scholar
-
7.X. A. Wu, Y. M. Zhao, N. J. Yu, J. Asian Nat. Prod. Res. 9(5), 437-441 (2007) CrossRef PubMed Google Scholar
-
8.(a) Cho, C. S. ; Oh, S. G. Tetrahedron Lett. 2006, 47(32), 5633-5636; (b) Climent, M. J. ; Corma, A. ; Iborra, S. ; MartinezSilvestre, S. ; Hernandez, J. C. ; Hungria, A. B. J. Catal. 2012, 292, 118-129. PubMed Google Scholar
-
9.C. S. Cho, W. X. Ren, J. Organomet. Chem. 694, 3215-3217 (2009) CrossRef PubMed Google Scholar
-
10.D. Bandyopadhyay, S. Mukherjee, R. R. Rodriguez, B. K. Banik, Molecules 15(6), 4207-4212 (2010) CrossRef PubMed Google Scholar
-
11.(a) El Khadem, H. ; Horton, D. ; Meshreki, M. H. ; Nashed, M. A. Carbohydr. Res. 1970, 13(2), 317-318; (b) Machell, G. ; Richards, G. N. J. Chem. Soc. 1960, 1938-1944; (c) Rowell, R. M. ; Green, J. Carbohydr. Res. 1970, 15(2), 197-203; (d) El Khadem, H. ; Horton, D. ; Meshreki, M. H. ; Nashed, M. A. Carbohydr. Res. 1971, 17(1), 183-192; (e) El Khadem, H. ; Horton, D. ; Meshreki, M. H. ; Nashed, M. A. Carbohydr. Res. 1972, 22(2), 381-389. PubMed Google Scholar
-
12.D. H. O'Donovan, I. Rozas, Tetrahedron Lett. 53(34), 4532-4535 (2012) CrossRef PubMed Google Scholar
-
13.(a) Seko, S. ; Komoto, I. J. Chem. Soc., Perkin Trans. 1 1998, (18), 2975-2976; (b) Seko, S. ; Tani, N. Tetrahedron Lett. 1998, 39(44), 8117-8120. PubMed Google Scholar
-
14.H. A. Goodwin, F. J. Lions, J. Am. Chem. Soc. 81, 6415 (1959) CrossRef PubMed Google Scholar
-
15.P. Ghosh, A. Mandal, Green Chem. Lett. Rev. 5(2), 127-134 (2012) CrossRef PubMed Google Scholar
-
16.A. Ohta, T. Watanabe, Y. Akita, M. Yoshida, S. Toda, T. Akamatsu, H. Ohno, A. Suzuki, J. Heterocycl. Chem. 19(5), 1061-1067 (1982) CrossRef PubMed Google Scholar
-
17.(a) Shaaban, M. ; Maskey, R. P. ; Wagner-Döbler, I. ; Laatsch, H. J. Nat. Prod. 2002, 65(11), 1660-1663; (b) Yu, D. Q. ; Yang, J. S., The Handbook of Analysis Chemistry, 2nd ed. Chemistry and Industry Publishing House: Beijing: 1999. PubMed Google Scholar
Copyright information
© The Author(s) 2013
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
