


Minor secoiridoid aglycones from the low-polarity part of the traditional Chinese herb: Swertia mileensis
Abstract
Eleven new secoiridoid aglycones involving unusual C9-skeleton:swerimilegenins A-F (1-6); bis-C9-skeleton:swerimilegenin G (7); and C10-skeleton:swerimilegenins H-K (8-11), as well as six known ones, were isolated from the low-polarity part of the traditional Chinese herb medicine Swertia mileensis. Their structures were determined by extensive spectroscopic data and X-ray diffraction. Biogenetically, swerimilegenin A (1) belonged to 10-nor-secoiridoid, and swerimilegenins B-F (2-6) were 1-nor-secoiridoids. Erythrocentaurin (12) and gentiogenal (15) showed moderate anti-HBV activity on HepG 2.2.15 cell line in vitro.Keywords
Swertia mileensis swerimilegenins secoiridoid aglycones anti-HBV activityIntroduction
Secoiridoids, are biogenetically derived from the cleavage of the cylcopentane ring of iridiods (generally between C-7 and C-8), and represent a large and still expanding group of monoterpenoids. Secoiridoids are widely present in mainly in Gentianales, Dipsacales, Cornales and Oleaceae.1, 2 They have attracted significant interest from scientists for their diverse bioactivities, and conferred a diverse array of actions such as antiviral, cardiovascular, choleretic, hypoglycemic, antiinflammatory and purgative activities, and chemotaxonomic significance.3 Secoiridoids consistently have a hemiacetal hydroxy group at C-1 position, which is unstable and apt to be glycosylated, thus most of the secoiridoids from natural resources are secoiridoid glycosides.4 According to the carbon numbers involved in the aglycone part, secoiridoids can be classified into three main types: Ⅰ) the normal skeleton with ten carbons; Ⅱ) skeletons with more than ten carbons in the aglycone part; and Ⅲ) skeletons with less than ten carbons. Presently, quite a few cases of secoiridoids other than C10 skeleton have been reported.5-7 Subsequently, the discovery of more secoiridoid aglycones with dissimilar skeletons will help to formulate a greater understanding of these compounds.
Previous investigations of Swertia mileensis ('Qing-YeDan' in Mandarin), which is a well-known traditional Chinese herb, endemic to the Yunnan province and documented in every edition of the Chinese Pharmacopoeia from 1977–2010, has shown that secoiridoid glycosides, xanthones and triterpenoids were their active constituents.8-10 As an ongoing search for anti-hepatitis B virus (anti-HBV) active compounds from natural resources, our recent investigation on the active part of S. mileensis resulted in a series of novel lactones, swerilactones A–O and swerilactosides A–C.11-17 From a biosynthetic perspective, swerilactones A and B (C18 skeleton) were derived from two C9 secoiridoids, and swerilactones H– K (C29 skeleton) were condensed by two C10 and one C9 secoiridoids. In addition, swerilactone G and swerilactosides A and B also contained the characteristic C9 secoiridoid moieties in their structures. The above analysis suggests that S. mileensis may be rich in secoiridoid aglycones, especially for the unusual C9 skeletons. Secoiridoids without glycosylation should be present in the low-polarity part of S. mileensis, however this part was ignored in the previous investigation, for its lowcontent and less-activity. Therefore, further investigation was focused on secoiridoid aglycones and was performed on the low-polarity part of S. mileensis, resulting in eleven new (1–11), as well as six pre-existing compounds (12–17). This paper will discuss their isolation, structural elucidation, and anti-HBV properties on HepG 2.2.15 cell line in vitro.

|
Results and Discussion
Compound 1 had the molecular formula of C9H12O4, which was determined by negative HRESIMS ([M – H]–, detected at m/z 183.0686, calcd for 183.0663), indicating four degrees of unsaturation. In the 13C NMR (DEPT) spectrum, three quaternary carbons, one methine and five methylenes, were observed. An α, β-unsaturated δ-lactone was proposed from the characteristic carbons at δC 163.0, 125.3, 149.5, 65.9 and 26.8, and HMBC correlations of H-7/C-5 and C-11, and H-6/C-4, which was in accordance with the UV (222 nm) and IR (1701 and 1631 cm−1) absorptions. Therefore, four residual carbons (three O-bearing methylenes and one methine) required an additional ring to fulfill the unsaturation. The connectivity of O-C(8)-C(9)-C(1)-O was constructed by the 1H-1H COSY experiment (H-8/H-9/H-1), of which C-9 was proposed to be attached with C-5 by HMBC correlations from H-1 and H-8 to C-5 (Figure 1). In HMBC spectrum, correlations from δH 4.43 and 4.29 (H-3) to δC 149.5 (C-5), 163.0 (C-11) and 66.5 (C-1) suggested that C-3 was directly linked to C-4 and further cyclized with C-1 by an oxygen atom. The configuration at C-9 was determined as R-form based on its optical rotation ([α]D18 – 12.4) and in comparison to the known compounds with the similar chiral center.18, 19 Thus, the structure of compound 1 was determined and named as swerimilegenin A.
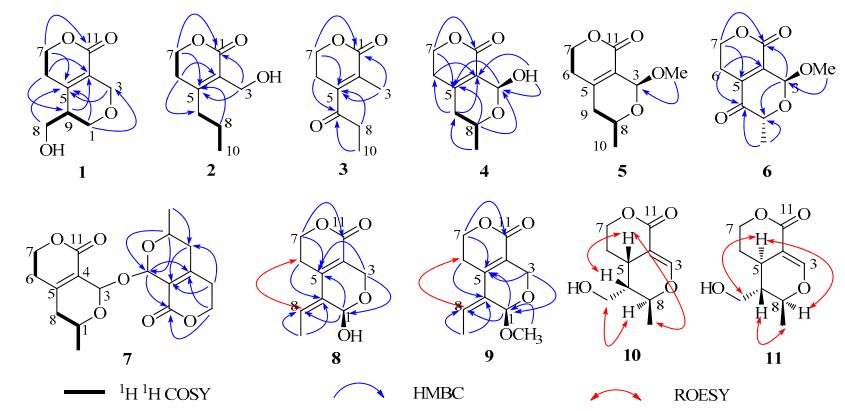
Selected 2D NMR correlations for compounds 1–11
Compound 2 was assigned the molecular formula of C9H14O3 by HREIMS which showed [M]+ at m/z 170.0944 (calcd. 170.0943), with three degrees of unsaturation. The existence of hydroxy and carbonyl groups was verified by IR absorptions at 3430 and 1711 cm–1. Nine carbons involving three quaternary ones, five methylenes and one methyl were detected in the 13C NMR (DEPT) spectrum. The characteristic signals at δC 166.2 (s, C-11), 156.8 (s, C-5), 126.2 (s, C-4), 65.5 (t, C-7) and 28.6 (t, C-6) enabled the α, β-unsaturated δ-lactone moiety. A propyl group was proposed at C-5 based on 1 H-1H COSY (H-10/H-8/H-9) and HMBC experiments (H-9/C-4, C-6 and H-8/C-5). One remaining oxygenated methylene (C-3) was recognized with a hydroxmethyl group and assigned at C-4 according to the HMBC correlations of H-3/C-4 and C-5. Thus, the structure of compound 2 was constructed.
Compound 3 possessed the molecular formula of C9H12O3 (four unsaturations) deduced from the pseudo-molecular ion peak of [M + Na]+ (m/z 191) in ESIMS and [M + H]+ (m/z 169.0866) in HRESIMS. The 1H and 13C NMR spectra of compound 3 were similar to those of 2, except that the hydroxymethyl (C-3) and methylene (C-9) in 2 were changed to be methyl and carbonyl in 3. Two groups of adjacent protons at δH 1.15 (t, J = 7.2 Hz, H-10) and 2.64 (q, J = 7.2 Hz, H-8) in 1H NMR spectrum, together with the carbons at δC 204.6 (s), 35.1 (t) and 7.4 (q) in 13C NMR (DEPT) spectrum suggested a propionyl group which was proposed at C-5 based on the HMBC correlations of H-8/C-5 and H-6/C-9. The remaining singlet methyl was assigned at C-4 by the long range correlations of H-3 with C-4, C-5 and C-11 in HMBC experiment.
Compound 4 was obtained as a hemiacetal analogue with the molecular formula of C9H12O4 by a positive HRESIMS experiment ([M + Na]+ at m/z 207.0624 and [2M + Na]+ at m/z 391.1245). The existence of hydroxy (3385 cm–1) and carbonyl (1686 cm–1) groups was specified in IR spectrum. A characteristic δ-lactone fragment was recognized from its 1H and 13C NMR data (Table 1). Besides the δ-lactone part, four residual carbons involving one di-oxygenated methine (C-3), one mono-oxygenated methine (C-8), one methylene (C-9), and one methyl (C-10), were proposed to occupy a cyclic pattern in agreement with the unsaturation degree. The proton signals at δH 1.17 (3H, d, J = 6.3 Hz, H-10), 4.28 (1H, m, H-8), 2.24 (1H, dd, J = 18.8, 3.6 Hz, H-9a) and 2.12 (1H, dd, J = 18.8, 10.8 Hz, H-9b) in 1H NMR spectrum, as well as the 1 H-1H COSY correlations of H-10/H-8/H-9 manifested the connectivity of CH3-CH(O-)-CH2-, which was allocated at C-5 by HMBC cross-peaks from H-8 to C-5, and from H-9 to C-4, C-5 and C-6. The hemiacetal methine (C-3) was determined to be directly linked with C-4 and further cyclized with C-8 by an O-bridge based on the HMBC experiment (H-3/C-5, C-8 and C-11; HO-3/C-4; H-8/C-3).
1H and 13C NMR data for compounds 1–5 (δ in ppm, J in Hz)
In order to determine the stereochemistry of C-3 and C-8, a ROESY experiment was performed, however the key correlation of H-8 or H-10 with H-3 was not detected. This phenomenon is frequently encountered in secoiridoids whose structure contains a six-member ring fused with a δ-lactone fragment. The connection of C(10)-C(8)-O-C(3)-H bears a stable W configuration which makes a homolateral H-3 and H-10 departure from each other. Meanwhile, the hydroxyl group at C-3 was apt to form intramolecule hydrogen bond with O=C(11), resulted in OH-3 apart from H-8. Therefore, no correlation of H-3/H-10 or OH-3/H-8 was observed in the ROESY experiment, despite having been located at the same orientation. In this case, the dihedral angles of H-9a/H-8 and H-9b/H-8 are about 56.8° and 176.4° according to the Chem 3D model calculation (Figure 2), which are consistent with the coupling constants of 3.6 Hz (JH-9a/H-8) and 10.8 Hz (JH-9b/H-8). In addition, compound 7, the dehydrated dimer of 4, was also obtained and unambiguously determined by X-ray single crystal diffraction. Compounds 4 and 7 had the almost identical NMR spectral data and similar specific rotation value (–2.06° for 4, about half of –6.65° for 7) indicating the same stereochemistry.
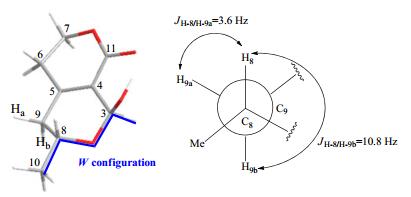
The W configuration in the Chem 3D model of compound 4
Compound 5 was proposed the molecular formula of C10H14O4 based on positive HRESIMS, 14 mass units (CH2) higher than swerimilegenin D (4). Its 1H and 13C NMR spectroscopic data were similar to those of 4 except for an additional methoxy group (δH 3.48, δC 55.7). With the aid of HMBC analysis, the methoxy group was deduced at C-3 by the correlation of OMe with C-3. Similar to swerimilegenin D, no correlation between H-3 (or OMe) and H-8 (or H-10) was detected in the ROESY experiment due to the W configuration. The stereocenters of C-3 and C-8 in compound 5 were deduced to be the same as those of 4 by comparing their coupling constant in 1H NMR spectrum and [α]D values.
Compound 6 was designated the molecular formula of C10H12O5 by positive HRESIMS ([M + Na]+, as detected at m/z 235.0586, calcd. for 235.0582). A methoxy group was speculated by the ion peak at m/z 181 (M+ – OMe) in the EIMS spectrum. Its 1H and 13C NMR data were similar to those of 5, except for the additional carbonyl group (196.6, s) instead of methylene [δC 36.8 (t, C-9)] in 5. The C(9)=O was evidenced based on the HMBC correlations of H-10/C-9, H-8/C-9 and C-5. Similar to compounds 4 and 5, neither H-8/H-3 nor Me-10/H-3 was detected in its ROESY spectrum due to the stable W configuration. The stereochemistry of compound 6 was proposed to be the same as compounds 4 and 5 by the coupling constant (JH-10/H-8 = 6.5 Hz) and optical rotation value ([α]D24 = – 4.0).
Compound 7 was determined with the molecular formula of C18H22O7 from positive HRESIMS (detected at m/z 373.1258). Only nine carbons and 11 protons were displayed in the 13C and 1H NMR spectra, indicating a symmetrical skeleton. Its 1H and 13C NMR spectral data were almost identical with those of 4, indicating the similar structures. The above analysis together with its molecular formula of C18H22O7 (18 mass units less than two molecules of 4) suggested that compound 7 should be the dehydrated dimer of 4, which was finally confirmed by extensive 2D NMR experiment and X-ray single crystal diffraction (Figure 3).
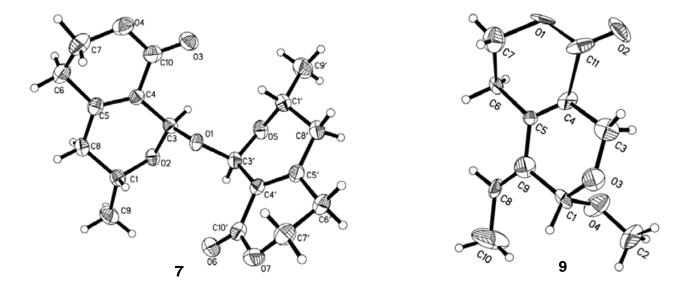
X-ray crystal structures of compounds 7 and 9
Compound 8 possessed the molecular formula of C10H12O4 in accordance with the ion peaks of M+ at m/z 196 in EIMS and [M + Na]+ at m/z 219.0641 in HRESIMS. The presence of OH and C=O groups was evidenced by the absorption bands at 3362 and 1699 cm–1 in IR spectrum. An obvious α, β-unsaturated δ-lactone fragment was revealed from its 1H and 13C NMR spectroscopic data (Table 2). In addition, one CH3CH=C moiety was proposed to be linked with C-5 and C-1 from the HMBC correlations of H-8 with C-5, H-6 with C-9 and H-1 with C-5 and C-8. The HMBC correlations of H-3 with C-4, C-5 and C-1 revealed the linkage of C(4)-C(3)-OC(1). The Z-form of the double bond between C-8 and C-9 was determined by the correlation of H-8 with H-6 in the ROESY spectrum.
1H and 13C NMR data for compounds 6–9 (δ in ppm, J in Hz)
Compound 9 had the molecular formula of C11H14O4 as a result of a positive HRESIMS experiment, with an additional CH2 moiety compared to compound 8. Its IR, UV and NMR spectroscopic data were similar to 8, except for an additional methoxy group. In HMBC spectrum, the correlations of OCH3/C-1 and H-1/OCH3 indicated that the methoxy group was located at C-1 position. Finally, its structure was determined by an X-ray single crystal diffraction experiment.
Compound 10 was proposed with a molecular formula of C10H14O4 by HREIMS at m/z 198.0897. The IR spectrum displayed the absorption bands at 3472, 1679, 1595 and 1404 cm –1 denoting the presence of OH, C=O and C=C groups. Its NMR spectroscopic data was close to that of dihydroepinaucledal (14), indicating a similar structure. From its 1H-1H COSY and HMBC experiments, compound 10 was established the same planar structure as 14. Therefore, their 1H and 13C NMR spectral differences (Table 3) might be ascribed to the stereochemical variation. In the ROESY spectrum, the correlations of H-5 with H-9 and H-10, and H-1 with H-8 suggested the same orientation of H-5 with H-9 and H-10, which display an obvious difference from the ROESY experiment of 14, in which the correlations of H-5/H-1 and H-10, and H-9/H-8 were observed (see Electronic Supplementary Material).
1H and 13C NMR data for compounds 10, 11 and dihydroepinaucledal (14) (δ in ppm, J in Hz)
Compound 11 possessed the molecular formula of C10H14O4 identical with swerimilegenin J from HRESIMS. The same planar structures of 11 with 10 and 14 were constructed based on extensive 1H-1H COSY and HMBC analysis. In the ROESY spectrum, the correlations of H-5 with H-1 and H-8, and H-9 with H-10 manifested the α-orientation of H-5, H-8 and hydroxymethyl (C-1), and the β-orientation of H-9 and Me-10.
The known dihydroepinaucledal (14) was determined by comparing its NMR data with that of a previous report20 and further confirmed by a 2D NMR experiment (see Electronic Supplementary Material). The other known compounds were determined as erythrocentaurin (12), 21 gentiolactone (13), 22 gentiogenal (15), 23 secostrychnosin (16)24 and angelone (17)5 by comparing their spectroscopic data to existing report data.
In order to evaluate their anti-HBV properties, namely inhibiting the secretions of hepatitis B surface antigen (HBsAg) and hepatitis B e antigen (HBeAg), as well as HBV DNA replication, the isolates were assayed on HepG 2.2.15 cell line in vitro (Table 4). Most of the tested compounds exhibited low activity and cytotoxicity at the highest tested concentration, except for erythrocentaurin and gentiogenal. Erythrocentaurin could inhibit the HBsAg secretion and HBV DNA replication with the IC50 values of 1.39 ± 0.54 and 0.96 ± 0.45 mM, respectively. Gentiogenal showed activity inhibiting the secretion of HBsAg and HBeAg with the IC50 values of 3.92 ± 0.82 and 2.99 ± 0.72 mM.
Anti-HBV activities of the isolates from S. mileensis (mM)
Previously, most of the secoiridoids isolated from Swertia plants were secoiridoid glycosides and always contained a C10 aglycone part. Interestingly, this investigation resulted in eleven new secoiridoid aglycones including unusual C9-skeleton (1–6), bis-C9-skeleton (7) and C10-skeleton (8–11) from the low-polarity part of S. mileensis. Structurally, compound 1 belonged to 10-nor-secoiridoid, and compounds 2–6 were attributed to 1-nor-secoiridoids, which further enriched the skeleton types of secoiridoids. It is concluded that diverse secoiridoid aglycones are contained in S. mileensis, although these compounds may be not corresponding to the anti-HBV activity in vitro.
Experimental Section
General Experimental Procedures. Melting points were measured on a SGWX-4B instrument (Shanghai Jingke, Shanghai, China). 1D and 2D NMR spectra were recorded on Bruker AV-400 NMR, DRX-500 or AVANCE Ⅲ-600 spectrometers (Bruker, Bremerhaven, Germany) corrected by deuterated solvent signals. MS data was collected on LCMSIT-TOF (Shimadzu, Kyoto, Japan) or VG Auto Spec-3000 (VG, Manchester, UK) spectrometers. IR (KBr) spectra was recorded on a Bruker Tensor 27 FT-IR (Bruker Optics GmbH, Ettlingen, Germany). UV data was collected on a Shimadzu UV-2401A spectrophotometer (Shimadzu, Kyoto, Japan). Optical rotations were collected on a Jasco model 1020 polarimeter (Horiba, Tokyo, Japan). Sephadex LH-20 (20–150 μm) was purchased from Pharmacia Fine Chemicals Co. Ltd. (Pharmacia, Uppsala, Sweden). Silica gel (200–300 mesh) for column chromatography was obtained from Qingdao Makall Chemical Company (Makall, Qingdao, China). CHP20P MCI gel (Mitsubishi Chemical Corporation, Tokyo, Japan) and Merck Lichrosorb RP-18 (Merck KGaA, Darmstadt, Germany) were applied for medium pressure liquid chromatographic (MPLC) preparation, which was performed on a Dr-Flash-S MPLC system (Lisui, Suzhou, China). Agilent Eclipse XDBC18 column (Agilent Technologies, Santa Clara, USA) was used for semi-preparation, which was conducted on a Waters 600-2695 HPLC instrument (Waters, Milford, MA, USA), equipped with a photodiode array detector (Waters 2996).
Plant Material. The whole plants of Swertia mileensis T. N. Ho et W. L. Shi were collected in Mile County, Yunnan Province, China, in November, 2008, and were identified by Prof. Li-Gong Lei. A voucher specimen (No. 2008-11-01) was deposited in the Laboratory of Antivirus and Natural Medicine Chemistry, Kunming Institute of Botany, CAS.
Extraction and Isolation. The air-dried and powered whole plant (5.0 kg) of S. mileensis was extracted with ethanol and condensed to provide a residue (1.3 kg). The residue was partitioned between water and petroleum ether (PE, 1 L × 2), ethyl acetate (EtOAc, 1 L × 3) and n-butanol (1 L × 3) successively. The EtOAc part (170 g) was chromatographed on silica gel column (2 kg, 110 mm id × 500 mm) eluted with CHCl3-MeOH to provide fractions A−J. Fraction A (29 g) was separated by silica gel column chromatography (CC, 300 g, 50 mm id × 240 mm) with an eluent of CHCl3-Me2CO system (0:100 → 50:50, v/v) to yield Frs. A1−A5.11 Frs. A1 and A2 were combined based on TLC detection which displayed many identical spots both under UV detector and treated with H2SO4 regent, and subjected to silica gel CC (200 g, 25 mm id × 600 mm) eluted with CHCl3/MeOH gradient (100:0, 95:5, 90:10 and 80:20, with 2 L, respectively), to obtain three fractions A1A–A1C. Fraction A1B (5 g) was separated by CHP20P MCI gel column chromatography (100.0 g, 25 mm id × 300 mm) on a MPLC system eluted with MeOH/H2O (from 20:80 to 80:20, 10 L) to yield seven sub-fractions (Frs. A1B-1 to A1B-7). Fr. A1B-1 (300 mg) was purified with a silica gel column (50 g, 25 mm id × 150 mm) eluting with PE/acetone (from 80:20 to 50:50, 800 mL) to afford compounds 3 (6 mg), 6 (8 mg), 12 (150 mg) and 15 (9 mg). Compounds 4 (5.0 mg), 5 (5.0 mg), 7 (6.0 mg), 8 (9.0 mg) and 9 (6.0 mg) were isolated from Fr. A1B-2 (100 mg) by Sephadex LH-20 (50.0 g, 14 mm id × 1450 mm, MeOH, 300 mL) and repeated silica gel CC (20 g, 14 mm id × 250 mm, PE/acetone = 80:20, 500 mL). Fr. A1B-3 (400 mg) was separated by RP-18 column (50 g, 25 mm id × 130 mm) eluted with MeOH/H2O (40:60, 1000 mL) on MPLC system and further purified by repeated silica gel CC to give compounds 1 (3 mg), 2 (3 mg) and 13 (6 mg). Compounds 17 (7 mg) and 16 (3 mg) were obtained from Fr. A1B-4 (60 mg) by repeated silica gel CC (20 g, 14 mm id × 250 mm) eluted with PE/acetone (70:30, 300 mL) and CHCl3/acetone (85:15, 300 mL) systems. Fr. A1B-5 (50 mg) was loaded on a HPLC apparatus using an Eclipse XDB-C18 column (94 mm id × 250 mm, 5 μm) and eluted with MeOH-H2O (25:75, flow rate = 3 mL/min) providing compounds 10 (4 mg), 11 (6 mg) and 14 (8 mg). The purity of all the isolates was proposed to be higher than 95% based on TLC (only one spot under UV radiation and iodine atmosphere) and NMR (smooth baseline without impurity peaks) methods.
Swerimilegenin A (1): white amorphous powder; [α]D18 – 12.4 (c 0.06, MeOH); UV (MeOH) λmax (log ε) 222 (3.54) nm; IR (KBr) νmax 3418, 2926, 1701, 1631, 1413, 1385, 1048 cm–1; for 1H and 13C NMR data, see Table 1; HRESIMS m/z 183.0686 [M – H]– (calcd. for C9H11O4, 183.0663).
Swerimilegenin B (2): white amorphous powder; UV (MeOH) λmax (log ε) 223 (3.82) nm; IR (KBr) νmax 3430, 2963, 1711, 1469, 1404, 1314, 1170, 1142, 1074, 1008 cm–1; 1H and 13C NMR data, see Table 1; HREIMS m/z 170.0944 M+ (calcd. for C9H14O3, 170.0943).
Swerimilegenin C (3): white amorphous powder; UV (MeOH) λmax (log ε) 229 (3.83) nm; IR (KBr) νmax 2939, 1720, 1403, 1309, 1173, 1130, 1072, 1130, 795 cm–1; 1H and 13C NMR data, see Table 1; ESIMS m/z 191 [M + Na]+; HRESIMS m/z 169.0866 [M + H]+ (calcd. for C9H13O3, 169.0864).
Swerimilegenin D (4): white amorphous powder; [α]D25 – 2.0 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 209 (3.89) nm; IR (KBr) νmax 3385, 2976, 1686, 1405, 1279, 1169, 1086, 1056, 1023, 986 cm–1; 1H and 13C NMR data, see Table 1; EIMS m/z 184 M+ (17), 183 (100), 167 (53), 140 (76), 125 (55), 94 (50); HRESIMS m/z 207.0624 [M + Na]+ (calcd. for C9H12O4Na, 207.0633).
Swerimilegenin E (5): white amorphous powder; [α]D22 – 6.1 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 207 (3.87) nm; IR (KBr) νmax 2908, 1712, 1410, 1279, 1150, 983, 966 cm–1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 221.0763 [M + Na]+ (calcd. for C10H14O4Na, 221.0784).
Swerimilegenin F (6): colorless gum; [α]D24 – 4.0 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 227 (3.87) nm; IR (KBr) νmax 2937, 1727, 1700, 1415, 1214, 1083, 1060, 975, 851 cm–1; 1H and 13C NMR data, see Table 2; EIMS m/z 181 (35), 168 (100), 151 (25), 140 (20), 110 (17), 82 (55); ESIMS m/z 235 [M + Na]+; HRESIMS m/z 235.0586 [M + Na]+ (calcd. for C10H12O5Na, 235.0582).
Swerimilegenin G (7): colorless cubic crystals; mp 228– 230 ℃; [α]D26 – 6.6 (c 0.3, MeOH/CHCl3); UV (MeOH) λmax (log ε) 210 (4.22) nm; IR (KBr) νmax 2976, 2936, 2908, 1721, 1707, 1473, 1414, 1323, 1281, 1156, 1060, 962, 809, 661, 561 cm–1; 1H and 13C NMR data, see Table 2; ESIMS m/z 373 [M + Na]+; HRESIMS m/z 373.1258 [M + Na]+ (calcd. for C18H22O7Na, 373.1263).
Swerimilegenin H (8): colorless gum; [α]D25 – 3.9 (c 0.4, MeOH); UV (MeOH) λmax (log ε) 273 (3.78), 237 (3.82) nm; IR (KBr) νmax 3362, 2954, 1699, 1637, 1419, 1286, 1064, 1030, 912, 773, 747 cm–1; 1H and 13C NMR data, see Table 2; EIMS m/z 196 M+ (10), 178 (90), 167 (20), 149 (24), 121 (30), 105 (26), 91 (100), 79 (25), 77 (45); ESIMS m/z 219 [M + Na]+; HRESIMS m/z 219.0641 [M + Na]+ (calcd. for C10H12O4Na, 219.0633).
Swerimilegenin I (9): colorless needles; mp 144–145 ℃; [α]D16 – 2.4 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 273 (4.21), 212 (3.66), 191 (3.90) nm; IR (KBr) νmax 2915, 1700, 1637, 1426, 1304, 1246, 1160, 1124, 1054, 1041 cm–1; 1H and 13C NMR data, see Table 2; ESIMS m/z 233 [M + Na]+; HRESIMS m/z 211.0964 [M + H]+ (calcd. for C11H15O4, 211.0970).
Swerimilegenin J (10): colorless gum; [α]D13 – 90.6 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 250 (3.98) nm; IR (KBr) νmax 3472, 2893, 1679, 1595, 1404, 1272, 1209, 1100, 1041, 962, 760 cm–1; 1H and 13C NMR data, see Table 3; EIMS m/z 199 [M + H]+ (40), 198 M+ (50), 168 (70), 127 (95), 123 (45), 97 (50), 57 (100); HREIMS m/z 198.0897 M+ (calcd. for C10H14O4, 198.0892).
Swerimilegenin K (11): colorless gum; [α]D19 + 2.2 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 249 (3.60) nm; IR (KBr) νmax 3431, 2921, 1695, 1614, 1409, 1277, 1110, 1053 cm–1; 1H and 13C NMR data, see Table 3; HRESIMS m/z 233.0567 [M + Cl]– (calcd. for C10H14O4Cl, 233.0586).
Crystallographic Data of Compounds 7 and 9: Swerimilegenin G (7): C18H22O7, MW = 350.14; monoclinic, space group p21; a = 9.5350(17) Å, b = 8.4098(15) Å, c = 21.881(4) Å, β = 101.266(2), V = 1720.8(5) Å3, Z = 7, d = 1.315 g/cm3, crystal dimensions 0.28 × 0.19 × 0.08 mm3 were used for measurement on a Rigaku D/MAX-3B X-ray diffractometer with a graphite monochromater, Mo Kα radiation. The total number of reflections measured was 10830, of which 4045, were observed, I > 2σ(I). Final indices: R1= 0.0523, wR2 = 0.1341.
Swerimilegenin I (9): C11H14O4, MW = 210.22; monoclinic, space group p21; a = 4.287(2) Å, b = 9.320(5) Å, c = 27.001(14) Å, β = 100.268(9), V = 1061.6(9) Å3, Z = 2, d = 1.315 g/cm3, crystal dimensions 0.20 × 0.13 × 0.06 mm3 were used for measurement on a Rigaku D/MAX-3B X-ray diffractometer with a graphite monochromater, Mo Kα radiation. The total number of reflections measured was 5809, of which 1253, were observed, I > 2σ(I). Final indices: R1= 0.1011, wR2 = 0.4433.
Anti-HBV Assay. The anti-HBV procedure was performed in accordance with our previous report.15 Tenofovir (Lot No. 200904009, purity > 97.6%) was used as the positive control, which was purchased from Jiangxi Chenyang Pharmaceutical Co. Ltd..
Electronic Supplementary Material
Supplementary material is available in the online version of this article at http://dx.doi.org/10.1007/s13659-013-0059-y and is accessible for authorized users.
Notes
Acknowledgments
This work was funded by the National Natural Science Foundation of China and Yunnan Province (U0832603), the National Science Foundation of China for Distinguished Young Scholars (81025023), the West Light Foundation of the Chinese Academy of Sciences, the International Foundation for Science (No. F/5202-1), and the Youth Innovation Promotion Association, CAS.
References
-
1.C. A. Boros, R. S. Stermitz, J. Nat. Prod. 53, 1055-1147 (1990) CrossRef PubMed Google Scholar
-
2.L. J. El-Naggar, J. L. Beal, J. Nat. Prod. 43, 649-707 (1980) CrossRef PubMed Google Scholar
-
3.B. Dinda, S. Debnath, Y. Harigaya, Chem. Pharm. Bull. 55, 159-222 (2007) CrossRef PubMed Google Scholar
-
4.S. R. Jensen, J. Schripsema, Gentianaceae-Systematic and Natural History:Gentianaceae-Systematics and Natural History[M]. Cambridge: Cambridge University, 2002, pp 573-630. PubMed Google Scholar
-
5.D. A. Mulholland, A. Langlois, M. Randrianarivelojosia, E. Derat, J. M. Nuzillard, Phytochem. Anal. 17, 87-90 (2006) CrossRef PubMed Google Scholar
-
6.K. He, Y. B. Ma, T. W. Cao, H. L. Wang, F. Q. Jiang, C. A. Geng, X. M. Zhang, J. J. Chen, Planta Med. 78, 814-820 (2012) CrossRef PubMed Google Scholar
-
7.H. L. Wang, C. A. Geng, Y. B. Ma, X. M. Zhang, J. J. Chen, Fitoterapia 89, 183-187 (2013) CrossRef PubMed Google Scholar
-
8.H. Kikuzaki, Y. Kawasaki, S. Kitamura, N. Nakatani, Planta Med. 62, 35-38 (1996) CrossRef PubMed Google Scholar
-
9.X. S. Li, Z. Y. Jiang, F. S. Wang, Y. B. Ma, X. M. Zhang, J. Chen, J. Chin. J. Chin. Mater. Med. 33, 2790-2793 (2008) PubMed Google Scholar
-
10.Y. Zhou, Y. T. Di, S. Gesang, S. L. Peng, L. S. Ding, Helv. Chim. Acta 89, 94-102 (2006) CrossRef PubMed Google Scholar
-
11.C. A. Geng, Z. Y. Jiang, Y. B. Ma, J. Luo, X. M. Zhang, H. L. Wang, Y. Shen, A. X. Zuo, J. Zhou, J. Chen, J. Org. Lett. 11, 4120-4123 (2009) CrossRef PubMed Google Scholar
-
12.C. A. Geng, X. M. Zhang, Y. Shen, A. X. Zuo, J. F. Liu, Y. B. Ma, J. Luo, J. Zhou, Z. Y. Jiang, J. Chen, J. Org. Lett. 11, 4838-4841 (2009) CrossRef PubMed Google Scholar
-
13.C. A. Geng, X. M. Zhang, Y. B. Ma, Z. Y. Jiang, J. Luo, J. Zhou, H. L. Wang, J. J. Chen, Tetrahedron Lett. 51, 2483-2485 (2010) CrossRef PubMed Google Scholar
-
14.C. A. Geng, X. M. Zhang, Y. B. Ma, Z. Y. Jiang, J. F. Liu, J. Zhou, J. J. Chen, J. Asian Nat. Prod. Res. 12, 542-548 (2010) CrossRef PubMed Google Scholar
-
15.C. A. Geng, L. J. Wang, X. M. Zhang, Y. B. Ma, X. Y. Huang, J. Luo, R. H. Guo, J. Zhou, Y. Shen, A. X. Zuo, Z. Y. Jiang, J. Chen, J. Chem. Eur. J. 17, 3893-3903 (2011) CrossRef PubMed Google Scholar
-
16.C. A. Geng, X. M. Zhang, Y. B. Ma, J. Luo, J. J. Chen, J. Nat. Prod. 74, 1822-1825 (2011) CrossRef PubMed Google Scholar
-
17.C. A. Geng, L. J. Wang, R. H. Guo, J. Chen, J. Mini. Rev. Med. Chem. 13, 749-776 (2013) CrossRef PubMed Google Scholar
-
18.J. J. Trost, B. S. Brown, E. J. McEachern, O. Kuhn, Chem. Eur. J. 9, 4442-4451 (2003) CrossRef PubMed Google Scholar
-
19.J. J. Verendel, J. Q. Li, X. Quan, B. Peters, T. G. Zhou, O. R. Gautun, T. Govender, P. G. Andersson, Chem. Eur. J. 18, 6507-6513 (2012) CrossRef PubMed Google Scholar
-
20.H. Takayama, H. Ishikawa, M. Kitajima, N. Aimi, B. M. Aji, Chem. Pharm. Bull. 52, 359-361 (2004) CrossRef PubMed Google Scholar
-
21.R. L. Nie, R. Y. He, Acta Bot. Yunnan. 6, 325-328 (1984) PubMed Google Scholar
-
22.J. H. Suhr, P. Arends, B. Jensen, Phytochemistry 17, 135-138 (1978) CrossRef PubMed Google Scholar
-
23.N. J. Vander, S. W. G. Vander, R. P. Labadie, Planta Med. 45, 161-162 (1982) PubMed Google Scholar
-
24.M. J. Cheng, I. L. Tsai, I. S. Chen, J. Chin. Chem. Soc. 48, 235-239 (2001) CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2017
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
