


Four new diterpenoids from Isodon eriocalyx var. laxiflora
Abstract
Four new diterpenoids, laxiflorins S-V (1-4), bearing four different types, and one known compound, laxiflorin O (5), were isolated from Isodon eriocalyx var. laxiflora. Compound 1 was the first example of ent-kauranoids bearing a unique C24 carbon framework and compound 4 was the first example of 3, 4-seco-ent-abietane diterpenoids from the Isodon genus. Their structures were determined by spectroscopic methods (UV, IR, MS, NMR).Keywords
Isodon eriocalyx var. laxiflora laxiflorins diterpenoid ent-kaurane ent-abietaneIntroduction
The Natural Products Library, is one of the primary sources for uncovering novel drug candidates, and is highly regarded in drug discovery.1-3 The construction of an ent-kaurane diterpenoids library was initiated by our group in 1976, and more than 1000 pure ent-kauranoids, which includes more than 700 novel discoveries, have been identified from Isodon genus. 4, 5
As one of the important plants of Isodon genus, I. eriocalyx var. laxiflora has been investigated phytochemically as it is a rich source of diterpenoids, such as 7, 20-epoxy-ent-kauranoids (laxiflorins H and I), 6 3, 20-epoxy-ent-kauranoids (laxiflorins J–M), 7, 8 6, 7-seco-ent-kauranoids (laxiflorins A–C), 9 6, 7:8, 15-seco-ent-kauranoids (laxiflorins F and G), 10 ent-abietanoids (laxiflorin N), 11 15, 16-seco-16, 17-dinor-ent-kauranoids (neolaxiflorins D–F)12, two unprecedented epimeric bishomoditerpene lactones with a unique C22 framework (laxiflorolides A and B), 13 and two unprecedented ent-kaurane diterpenoids (neolaxiflorins A and B).14 Our further investigation of this plant led to the isolation of four novel ones, laxiflorins S–V (1–4), and one known compound, laxiflorin O (5)15 (Figure 1). These new compounds could be classified into 4 different types as below: tetrahomo-7, 20-epoxy-ent-kauranoid (1), 7, 20-epoxy-ent-kauranoid (2), 15, 16-seco-16, 17-dinor-entkauranoid (3), and 3, 4-seco-ent-abietane diterpenoid (4), respectively. In addition, compound 1 was the first example of ent-kauranoids bearing a unique C24 carbon framework; compound 3 was the second 15, 16-seco-16, 17-dinor-entkauranoid obtained from the Isodon genus; compound 4 was the first example of 3, 4-seco-ent-abietane diterpenoid obtained from the Isodon genus plants. In this paper, we report the isolation and structure elucidation of compounds 1–5.
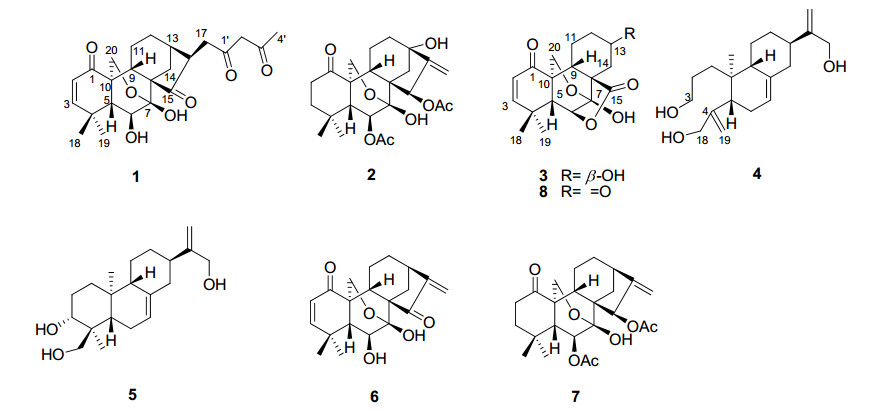
Chemical structures of compounds 1–8
Results and Discussion
Laxiflorin S (1) was obtained as an amorphous powder. The molecular formula C24H30O7, has ten degrees of unsaturation, was established based on HREIMS ([M]+, 430.2003; calcd for C24H30O7, 430.1992) and NMR spectroscopy (Tables 1 and 2). The IR spectrum of 1 indicated the presence of hydroxy groups (3480 cm–1) and two types of carbonyl group for saturated ketone (1741 cm–1), and conjugated ketone (1648 cm–1), respectively.
1H NMR data (δ in ppm, J in Hz, C5D5N) of compounds 1–4
13C NMR data (δ in ppm, C5D5N) of compounds 1–4
The 1H NMR spectrum (Tables 1 and 2) showed resonances attributed to an α, β-unsaturated ketone moiety δH 5.70 (1H, ABd, J = 10.1 Hz, 6.41, 1H, ABd, J = 10.1 Hz), two tertiary methyls at δH 1.08 (3H, s) and δH 0.80 (3H, s), and quaternary methyl at δH 1.75 (3H, s). In addition, the spectrum showed resonances due to a characteristic AB methylene group at (δH 4.21 and 3.82, each 1H, d, J = 9.8 Hz), together with an oxygenated methine at δH 3.97 (dd, J = 11.3, 7.4 Hz). The analysis of the 13C NMR and DEPT spectra (Table 2) revealed the presence of 24 carbons, which were assigned as three methyls (δC 29.9, δC 24.1, and δC 29.7), six methylenes (one oxygenated at δC 65.6), seven methines (one oxygenated at δC 73.5 and two olefinic groups at δC 127.4 and δC 160.7), and eight quaternary carbons (one hemiacetal at δC 96.3 and four carbonyls at δC 197.4, δC 225.2, δC 207.5, and δC 206.1), which suggested that 1 is a highly oxygenated diterpenoid with a C24 skeleton similar to the ent-kaurane skeletons previously reported.4, 9 Comparison of the NMR spectral data of 1 with those of eriocalyxin B (6)16 revealed that 1 was consistent with a 7, 20-epoxy-ent-kaurane similar to 6 by reducing the carboncarbon double bond between C-16 and C-17 of 6 and then adding a butyl-1, 3-dione group at C-17.
The HMBC spectrum of 1 showed obvious correlations from the geminal methyls Me-18 and Me-19 to C-3, C-4, and C-5, and from Me-4' to C-2' and C-3'. In addition, the AB methylene H2-20 showed HMBC correlations with C-1, C-5, C-7, C-9, and C-10 respectively. Other HMBC correlations were noted between the AB methine H-2 and C-1, C-3, C-4, and C-10, and between H-3 and C-1, C-4, C-5, C-18, and C-19, between H-6 (δH 3.97) and C-4, C-5, C-7, C-8, and C-10, and between H-17 and C-13, C-15, C-16, C-1', and C-2'. These observed HMBC correlations coupled with three spin systems (i.e., (CHCH, H-2/H-3), (CHCH, H-5/H-6), and (CHCH2CH2CH(CHCH2)CH2, H-9/H2-11/H2-12/H-13(/H-16/H2-17))/H2-14)) established by 1H-1H COSY correlations and HSQC spectra suggested the gross structure of compound 1 (Figure 2).
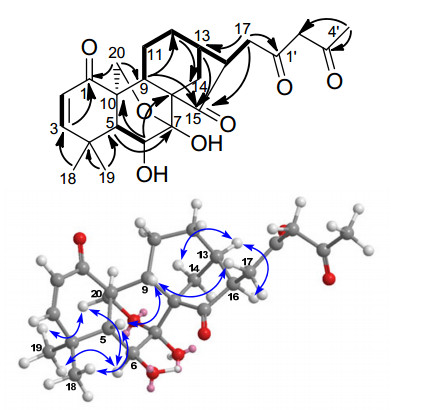
1H-1H COSY (▂), selected HMBC (H→C) and key ROESY (
In the ROESY spectrum of compound 1, the NOE correlations for Me-19/H-6, H-6/H2-20, H-20a/H-14a, H-13/H-16, and H-16/H2-14 suggested that H-6, H-13, C-14, H-16, Me-19, and C-20 adopted the same α-orientation. The cross-peaks observed between H-5/H-9, H-5/Me-18, and H-9/H2-17 in the ROESY spectrum demonstrated that H-5, H-9, C-17, and Me-18 all possessed the same β-orientation. (Figure 2) Therefore, compound 2 was 6β, 7β-dihydroxy-17-(butyl 1', 3'-dione)-7α, 20-epoxy-ent-kaur-2-en-1, 15-dione, and named laxiflorin S.
The HREIMS (m/z 448.2087, [M]+; calcd for C24H32O8, 448.2097) of compound 2 suggested a molecular formula of C24H32O8, with nine degrees of unsaturation. The 1H and 13C NMR data of 2 (Tables 1 and 2) were consistent with a 7, 20-epoxy-ent-kaurane, similar to 7β-hydroxy-6β, 15β-diacetoxy-7, 20-epoxy-ent-kaur-16-en-1-one (7)17. The most notable difference is that the C-13 methine group in 7 was substituted by a hydroxy group in 2. This difference was supported by HMBC correlations n gcfgnffrom H-15 (δH 6.40, s) and H2-17 (δH 5.81, d, J = 2.2 Hz and δH 5.44, d, J = 2.2 Hz) to C-13 (δC 75.4, s). The ROESY cross peaks correlations indicated that compounds 2 and 7 had the same relative configuration. Consequently, compound 2 was 7β, 13α-dihydroxy-6β, 15β-diacetoxy-7, 20-epoxy-ent-kaur-16-en-1-one, and it was named laxiflorin T.
The absolute configuration of neolaxiflorin D (8), a 15, 16-seco-16, 17-dinor-ent-kaurane diterpenoid isolated from the title plant12, was confirmed by single-crystal X-ray diffraction using anomalous scattering of CuKα radiation (CCDC 861510).18, 19 Using HREIMS (m/z 334.1416; calcd for C18H22O6, 334.1416), the molecular formula of laxiflorin U (3) was determined to be C18H22O6, indicating eight degrees of unsaturation. Its 1H and 13C NMR data (Tables 1 and 2) indicated that it was structurally very similar to neolaxiflorin D (8).12 The most notable difference was that the C-13 carbonyl group in compound 8 (δC 208.7, s)12 changed into hydroxy group in compound 3 (δC 63.8, d). This structural assignment was supported by the observed HMBC correlations between H-13 (δH 5.30, m) with C-8, C-11, C-12, and C-14, coupled with three spin systems (i.e., (CHCH, H-2/H-3), (CHCH, H-5/H-6), and (CHCH2CH2CHCH2, H-9/H2-11/H2-12/H-13/H2-14)) established by 1H-1H COSY correlations and HSQC spectra. Thus compound 3 was 7, 20-epoxy-7β, 13β-dihydroxy-1-oxo-15, 16-seco-16, 17-dinor-ent-kaur-2-en-6β, 15-olide, and it was named laxiflorin U.
Laxiflorin O (5) was an ent-abietane diterpenoid isolated from I. eriocalyx var. laxiflora, and its structure was confirmed by NMR and single-crystal X-ray diffraction in 2002.15 The molecular formula of compound 4 was determined to be C20H32O3 based on HREIMS (m/z 320.2343, [M]+; calcd. for C20H32O3, 320.2351). A comparison of its corresponding 1H and 13C NMR data (Tables 1 and 2) with those of laxiflorin O (5)15 indicates that compound 4 was the 3, 4-cleavage derivative of 5. The structural difference between these two compounds are that the oxygenated methine group at C-3 (δC 73.9, d) in 5 changed into a hydroxymethyl in 4 (δC 62.9, t), and saturated quaternary carbon at C-4 (δC 38.3, s) and C-19 (δC 23.5, q) in 5 changed into a double bond at C-4 (δC 153.5, s) and C-19 (δC 110.6, q) in 4. The HMBC correlations (Figure 3) of H2-18 (δH 4.51, d, J = 11.6 Hz and δH 4.40, d, J = 11.6 Hz) with C-4, C-5, and C-19 and of H2-19 (δH 5.70, br. s and δH 5.14, br. s) with C-4, C-5, and C-18, coupled with 1H-1H COSY correlations (Figure 3) (for the observed proton spin systems, H-9/H2-11/H2-12/H-13/H2-14, H2-1/H2-2/H2-3, and H-5/H2-6/H-7) confirmed this result. Based on detailed analysis of ROESY data, the relative configuration of the stereogenic centers in compound 4 were determined to be the same as those in laxiflorin O (5). The correlations from Me-20 to H-11α; from H-11α to H-13α; from H-5 to H-9, and from H-9 to H-11β was observed in the ROESY spectrum, and thus H-13 and Me-20 were assumed to be α-oriented, and of H-5 and H-9 were assumed to be β-oriented. Therefore, compound 4 was 3, 16, 18-trihydroxy-3, 4-seco-ent-abieta-4(19), 7(8), 15-triene, and it was named as laxiflorin V.
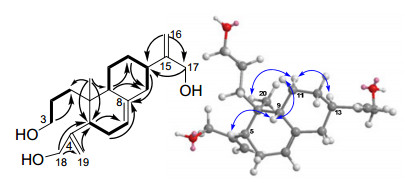
1H-1H COSY (▂), selected HMBC (H→C) and key ROESY (
In summary, four new diterpenoids belonging to four different types as: tetrahomo-7, 20-epoxy-ent-kauranoid, 7, 20-epoxy-ent-kauranoid, 15, 16-seco-16, 17-dinor-ent-kauranoid, and 3, 4-seco-ent-abietane diterpenoid, have been isolated from I. eriocalyx var. laxiflora collected in the south-west of China. Their structures were determined by analyses of 1D and 2D NMR spectroscopic data.
Additionally, compound 1 was the first example of ent-kauranoids bearing a unique C24 carbon framework and compound 4 was the first example of 3, 4-seco-ent-abietane diterpenoid obtained from the Isodon genus plants.
Experimental Section
General Experimental Procedures. Optical rotations were measured with a JASCO DIP-370 digital polarimeter. UV data were obtained on a Shimadzu UV-2401A spectrophotometer. A BioRad FtS-135 spectrophotometer was used for scanning IR spectroscopy with KBr pellets. 1D and 2D NMR spectra were recorded on DRX-500 and DRX-600 spectrometers. Unless otherwise noted, the chemical shifts (δ) are expressed in ppm with respect to the solvent signals. HREIMS was performed on a VG Autospec-3000 spectrometer at 70 eV. Column chromatography was performed with silica gel (100–200 mesh; Qingdao Marine Chemical, Inc., Qingdao, China). PPLC was performed on a Shimadzu LC-8A preparative liquid chromatograph with a Shimadzu PRC-ODS (K) column. The fractions were monitored by TLC, and the spots were visualized by heating silica gel plates sprayed with 8% H2SO4 in EtOH. All of the solvents including petroleum ether (60–90 ℃) were distilled prior to use.
Plant Material. The leaves of Isodon eriocalyx var. laxiflora were collected from Yunnan Province, China in September 2009. Voucher specimens (KIB20080028) were deposited at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences and were identified by Prof. Xi-Wen Li.
Extraction and Isolation. The air-dried leaves of Isodon eriocalyx var. laxiflora (10 Kg) were extracted with 70% aqueous Me2CO (3 × 40 L, 2 days each) at room temperature. The solvent was evaporated in vacuo to afford a crude extract, which was suspended in H2O and then successively extracted with EtOAc and n-BuOH. The EtOAcsoluble fraction (600 g) was decolorized on an MCI gel with 90:10 MeOH:H2O to obtain a yellow gum (427.5 g). The gum was purified by CC on silica gel column with a CHCl3:Me2CO gradient system consisting of 1:0, 9:1, 8:2, 7:3, 6:4, and 1:1 to yield six main fractions (A–F). Fraction C (CHCl3:Me2CO, 8:2; 30 g) was subjected to repeated chromatography over silica gel (CHCl3:MeOH, from 90:1, 60:1, to 30:1) to yield fractions C1–C3. Fraction C2 (CHCl3:MeOH, 60:1, 15 g) was separated by RP-18 CC (MeOH:H2O, 15:85 to 1:0) to afford C2/1–C2/4. Compound 1 (10 mg) was isolated from fraction C2/3 (500 mg) using PPLC (MeCN:H2O, 35:65, 15 mL/min) to achieve this separation. Fraction C3 (CHCl3: MeOH, 5 g) was eluted with RP-18 CC (MeOH:H2O, 10:90 to 1:0) yielding subfractions C3/1–C3/3. Subfraction C3/1 (1.06 g) was fractionated by repeated CC, first on silica gel column with a gradient elution with CHCl3: isopropyl alcohol (60:1 to 20:1) to yield fractions C3/1/1– C3/1/3. Subsequently, fraction C3/1/1 (250 mg) was purified using RP-18 CC (MeOH:H2O, 45:60) to give 4 (6 mg).
Fraction D (CHCl3:Me2CO 7:3, 50 g) was eluted with CHCl3:MeOH (30:1, 20:1, and 10:1) yielding sub-fractions D1–D3. Sub-fraction D1 (CHCl3:MeOH, 30:1, 20 g, ) was fractionated by repeated CC, first on RP-18 with a gradient elution with MeOH:H2O (2:8 to 1:0) to yield fractions D1/1– D1/8. Subsequently, fraction D1/3 (2.27 g) was purified using a silica gel column (CHCl3: MeOH, 50:1 to 10:1) to give subfractions D1/3/1–D1/3/8. Sub-fraction D1/3/3 was purified by PPLC (MeOH:H2O, 40:60, 20 mL/min) to yield 5 (19 mg). Sub-fraction D2 (10 g, CHCl3:MeOH, 20:1) was fractionated by repeated CC, first on RP-18 with gradient elution with MeOH:H2O (2:8 to 1:0) to yield fractions D2/1-D2/5. Subsequently, fraction D2/2 (0.87 g) was purified using a silica gel column (CHCl3: isopropyl alcohol, 30:1 to 10:1) to afford subfractions D2/2/1 (180 mg), D2/2/2 (105 mg), D2/2/3 (150 mg), and D2/2/4 (120 mg). Sub-fraction D2/2/3 was purified by RP-18 CC (MeOH:H2O, 40:60) to yield 2 (2 mg). Sub-fraction D3 (10:1 CHCl3:MeOH, 6 g) was purified by CC on RP-18 (15:85-1:0, MeOH-H2O) to yield fractions D3/1–D3/6. Subsequently, fraction D3/3 (1.26 g) was purified by CC on silica gel column (30:1-10:1 CHCl3:isopropyl alcohol) to yield subfractions D3/3/1 (750 mg), D3/3/2 (85 mg), and D3/3/3 (120 mg). compound 3 (6 mg) was precipitated from sub-fraction D3/3/1 by subsequent silica gel CC (20:1 CHCl3:MeOH) and RP-18 (40:60 MeOH:H2O).
Laxiflorin S (1): white, amorphous powder. [α]D21.0 – 53.1 (c 0.10, MeOH). UV (MeOH) λmax (log ε): 199.2 (3.0), 237.2 (3.3) nm; IR (KBr) νmax 3480, 2977, 2961, 2851, 1741, 1647, 1455, 1434, 1371, 1259, 1221, 1033 cm–1; For 1H and 13C spectroscopic data, see Tables 1 and 2; EIMS: m/z 430 [M]+; HREIMS [M]+ m/z 430.2003 (calcd for C24H30O7, 430.1992).
Laxiflorin T (2): white, amorphous powder. [α]D24.5+ 1.2 (c 0.10, MeOH). UV (MeOH) λmax (log ε): 202.6 (2.7) nm; IR (KBr) νmax 3521, 2958, 2933, 1740, 1726, 1696, 1428, 1379, 1285, 1116, 1040 cm–1; For 1H and 13C spectroscopic data, see Tables 1 and 2; Positive ESIMS: m/z 471 [M + Na]+; HREIMS [M]+ m/z 448.2087 (calcd for C24H32O8, 448.2097).
Laxiflorin U (3): white power. [α] 22.0 D + 21.6 (c 0.10, MeOH). UV (MeOH) λmax (log ε): 227 (3.03) nm. IR (KBr) νmax 3402, 2961, 2924, 1780, 1723, 1700, 1671, 1630, 1379, 1228, 1072 cm–1; For 1H and 13C spectroscopic data, see Tables 1 and 2; EIMS: m/z 334 [M]+; HREIMS [M]+ m/z 334.1416 (calcd for C18H22O6, 334.1416).
Laxiflorin V (4): white, amorphous powder. [α]D18.3– 20.3 (c 0.10, MeOH). UV (MeOH) λmax (log ε): 203.0 (3.1) nm; IR (KBr) νmax 3440, 2926, 2867, 1708, 1690, 1641, 1459, 1449, 1411, 1378, 1307, 1237, 1052 cm–1; For 1H and 13C spectroscopic data, see Tables 1 and 2; Positive ESIMS: m/z 343 [M + Na]+; HREIMS [M]+ m/z 320.2343 (calcd for C20H32O3, 320.2351).
Notes
Electronic Supplementary Material
Supplementary material is available in the online version of this article at http://dx.doi.org/10.1007/s13659-013-0057-0 and is accessible for authorized users.
Acknowledgements
The authors are grateful to Prof. Xi-Wen Li of the Kunming Institute of Botany, Chinese Academy of Sciences, for the identification of the plant. This project was supported financially by the National Natural Science Foundation of China (No. 81172939), the West Light Foundation of the Chinese Academy of Sciences (J.X. Pu), the Major State Basic Research Development Program of China (No. 2009CB522300), the reservation talent project of Yunnan Province (2011CI043 to J.X. Pu), the Science and Technology Program of Yunnan Province (Nos. 2008IF010), and the Major Direction Projection Foundation of CAS Intellectual Innovation Project (No. KSCX2-EW-J-24 to J.X. Pu).
References
-
1.J. W. Li, J. C. Vederas, Science 325, 161-165 (2009) CrossRef PubMed Google Scholar
-
2.S. Dandapani, L. A. Marcaurelle, Nat. Chem. Biol. 6, 861-863 (2010) CrossRef PubMed Google Scholar
-
3.J. Medina-Franco, Drug Des 1, e105 (2012) PubMed Google Scholar
-
4.H. D. Sun, S. X. Huang, Q. B. Han, Nat. Prod. Rep. 23, 673-698 (2006) CrossRef PubMed Google Scholar
-
5.R. Zhan, X. Du, J. Su, X. N. Li, W. G. Wang, C. Q. Liang, J. H. Yang, Y. Li, J. X. Pu, H. D. Sun, Nat. Prod. Bioprospect. 1, 116-120 (2011) CrossRef PubMed Google Scholar
-
6.X. M. Niu, S. H. Li, M. L. Li, Q. S. Zhao, S. X. Mei, Z. Na, S. J. Wang, Z. W. Lin, H. D. Sun, Planta Med 68(6), 528-533 (2002) CrossRef PubMed Google Scholar
-
7.W. G. Wang, H. Y. Wu, X. Du, J. M. Yan, Y. Li, J. X. Pu, H. D. Sun, Chin. J. Chem. 30, 1226-1230 (2012) CrossRef PubMed Google Scholar
-
8.X. M. Niu, S. H. Li, S. X. Mei, Z. Na, Q. S. Zhao, Z. W. Lin, H. D. Sun, J. Nat. Prod. 65, 1892-1896 (2002) CrossRef PubMed Google Scholar
-
9.H. D. Sun, Z. W. Lin, F. D. Niu, P. Q. Shen, L. T. Pan, L. Z. Lin, G. A. Cordell, Phytochemistry 38, 1451-1455 (1995) CrossRef PubMed Google Scholar
-
10.X. Niu, S. Li, Q. Zhao, Z. Lin, H. Sun, Y. Lu, C. Wang, Q. Zheng, Tetrahedron Lett. 43, 661-664 (2002) CrossRef PubMed Google Scholar
-
11.X. Niu, S. Li, Q. Zhao, S. Mei, Z. Lin, H. Sun, Y. Lu, C. Wang, Q. Zheng, Helv. Chim. Acta 86, 299-306 (2003) CrossRef PubMed Google Scholar
-
12.W. G. Wang, X. N. Li, X. Du, K. Dong, W. Zhao, H. Y. Wu, L. M. Kong, Y. Li, J. X. Pu, H. D. Sun, , Tetrahedron Lett. 53, 2777-2781 (2012) CrossRef PubMed Google Scholar
-
13.W. G. Wang, X. N. Li, X. Du, H. Y. Wu, X. Liu, J. Su, Y. Li, J. X. Pu, H. D. Sun, J. Nat. Prod. 75, 1102-1107 (2012) CrossRef PubMed Google Scholar
-
14.W. G. Wang, X. Du, X. N. Li, H. Y. Wu, X. Liu, S. Z. Shang, R. Zhan, C. Q. Liang, L. M. Kong, Y. Li, J. X. Pu, H. D. Sun, Org. Lett. 14, 302-305 (2012) CrossRef PubMed Google Scholar
-
15.X. M. Niu, S. H. Li, M. L. Li, Q. S. Zhao, S. X. Mei, Z. Na, S. J. Wang, Z. W. Lin, H. D. Sun, Planta Med. 68, 528-533 (2002) CrossRef PubMed Google Scholar
-
16.P. Shen, H. Sun, Z. Lin, Yunnan Zhiwu Yanjiu 8, 163-166 (1986) PubMed Google Scholar
-
17.E. Fujita, T. Fujita, M. Shibuya, Chem. Commun, 148-149 (1967) PubMed Google Scholar
-
18.H. Flack, G. Bernardinelli, Acta Crystallogr., Sect. A 55, 908-915 (1999) CrossRef PubMed Google Scholar
-
19.H. D. Flack, Acta Crystallogr., Sect. A 39, 876-881 (1983) CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2013
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
