


ent-Kaurane diterpenoids from the plant Wedelia trilobata
Abstract
Four new ent-kaurane diterpenoids, namely, 3α-tigloyloxypterokaurene L3 (1), ent-17-hydroxy-kaura-9(11), 15-dien-19-oic acid (2), and wedelobatins A (3) and B (4), together with 11 known ent-kaurane diterpenoids (5-15), were isolated from the ethanol extract of Wedelia trilobata. All the structures of 1-15 were elucidated on the basis of spectroscopic studies.Keywords
ent-kaurane diterpenoids Wedelia trilobata phytochemical investigationIntroduction
Wedelia trilobata has been used as a traditional herbal medicine for the treatment of fever and malaria in Vietnam and also for the treatment of backache, stubborn wounds, sores, and arthritic pain in the Caribbean and Central America.1, 2 Previous phytochemical studies showed that the plants of this genus are a rich source of ent-kaurane diterpenoids.3-5
As part of our efforts to assemble a large scale natural product library with thousands of compounds derived from plants and micro-organisms, phytochemical investigation on W. trilobata led to the isolation of four new ent-kaurane diterpenoids, namely, 3α-tigloyloxypterokaurene L3 (1), ent-17-hydroxykaura-9(11), 15-dien-19-oic acid (2), and wedelobatins A (3) and B (4), together with 11 ent-kaurane derivatives, grandiflorenic acid (5), 6 pterokaurene L3 (6), 7 3α-cinnamoyloxy-pterokaurene L3 (7), 3 ent-3β-cinnamoyloxykaur-16-en-19-oic acid (8), 4 grandifloric acid (9), 8 ent-17-hydroxykaur-15-en-19-oic acid (10), 3 3α-angeloyloxypterokaurene L3 (11), 3 ent-3β-tigloyloxykaur-16-en-19-oic acid (12), 10 ent-3β-angeloyloxykaur-16-en-19-oic acid (13), 3, 10 12α-methoxygrandiflorenic acid (14), 11 and 12α-hydroxygrandiflorenic acid (15).12 This paper herein describes the isolation and structural elucidation of these new compounds.

|
Results and Discussion
3α-Tigloyloxypterokaurene L3 (1), was obtained as a white amorphous powder, with its molecular formula determined as C25H36O5 on the basis of HREIMS, showing a molecular ion peak at m/z 416.2554 (calcd for C25H36O5, 416.2563). The IR spectrum revealed absorption bands of hydroxyl (3513 cm-1), carbonyl (1701 cm-1), and double bond (1652 cm-1) groups. In the 1H NMR spectrum (Table 1), the downfield olefinic proton at δH 6.86 (br. q, J = 7.1 Hz) and two methyl signals at δH 1.77 (br. d, J = 7.1 Hz) and 1.82 (br. s), was indicated by the presence of a tigloyloxy group in 1.10 Apart from the five carbon signals assigned to the tigloyloxy group (δC 167.7, 128.8, 137.2, 14.4, and 12.0), 10 the 13C NMR (DEPT) spectrum (Table 1) of 1 also exhibited 20 carbons composed of two methyls, nine methylenes, three methines (one oxygenated), and six quaternary carbons, which were consistent with a skeleton of an ent-kauranoid.7 In particular, the NMR spectroscopic features of 1 are similar to those of 6 (pterokaurene L3), which only differed in the appearance of a tigloyloxy group at C-3 in 1. It was also confirmed by the chemical shift value of C-3 (δC 78.7, CH), C-9 (δC 77.3, C) and the HMBC correlations (Figure 1) from H-3 (δH 4.60, dd, J = 12.1, 4.5 Hz) to C-1′ (δC 167.7, C), C-1 (δC 30.6, CH2), and C-18 (δC 24.1, CH3) as well as the correlations from Me-20, H-12, and H-15 to C-9, and from the methyl at C-4 (Me-18) to a downfield quaternary carbon (C-19) at δC 180.2. The β-orientation of the hydroxy group at C-9 in 1 was based on the downfield shift of H-15β (δH 2.70) and upfield shift of C-15 (δC 43.7) for the γ-steric compression effect in 1 from the hydroxyl group at C-9 to H-15β as evidenced in pterokaurene L3 (6).7 Furthermore, the ROESY correlations (Figure 2) of H-3 with H-5 and Me-18 suggested that the tigloyloxy was α-orientated. Consequently, the structure of 1 was finally determined as ent-3β-tigloyloxy-9α-hydroxykaur-16-en-19-oic acid, and given the name as 3α-tigloyloxypterokaurene L3.
NMR data of compounds 1 and 2 (13C NMR, 100 MHz, 1H NMR, 600 MHz; CDCl3)
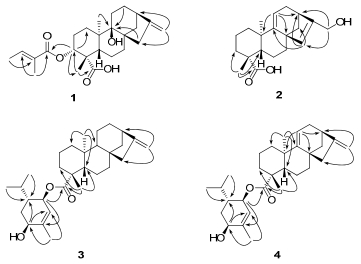
Key HMBC correlations of compounds 1–4
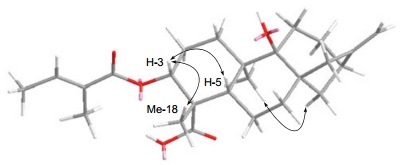
Key ROESY correlations of compound 1
ent-17-Hydroxykaura-9(11), 15-dien-19-oic acid (2), was obtained as a white, amorphous powder, with a molecular formula of C20H28O3 on the basis of HREIMS, showing a molecular ion peak at m/z 316.2028 (calcd for C20H28O3, 316.2038). The IR spectrum indicated the presence of hydroxyl (3427 cm-1), carbonyl (1693 cm-1), and double bond (1639 cm-1) groups. The 13C NMR (DEPT) spectrum (Table 1) revealed 20 carbons including three sp3 quaternary carbons, three sp2 quaternary carbons (one carboxylic acid carbonyl), two sp3 methines, two sp2 methines, eight sp3 methylenes (one oxygenated), and two methyl groups. Its 1H NMR spectrum (Table 1) showed two olefinic protons at δH 6.01 (s) and 5.00 (dd, J = 3.7, 3.1 Hz), two AB double doublets assigned to the protons of a hydroxymethyl group at δH 4.25 (dd, J = 14.1, 1.4 Hz) and 4.21 (dd, J = 14.1, 0.9 Hz), and two methyl signals at δH 1.24 and 1.01 (each 3H, s). These spectroscopic features suggested that the structure of 2 was similar to that of 10 (ent-17-hydroxykaur-15-en-19-oic acid), 3 and only differed in appearance as a double bond between C-9 (δC 157.2, C) and C-11 (δC 113.5, CH) in 2. It was confirmed by, the HMBC correlations from H-11 (δH 5.00, dd, J = 3.7, 3.1 Hz) to C-8 (δC 45.9, C), C-10 (δC 38.1, C), and C-13 (δC 38.8, CH), as shown in Figure 1. The α-orientation of the carboxylic acid group at C-4 was inferred from the 13C NMR chemical shift of the methyl group at C-4 by comparing those of related ent-kaurane diterpenoids, in which the methyl group with β-orientation resulted in resonance of approximately δC 29, as opposed to resonance of approximately δC 16 when the methyl group was in the α-orientation.7, 13, 14 Accordingly, the structure of compound 2 was elucidated as ent-17-hydroxykaura-9(11), 15-dien-19-oic acid.
Wedelobatin A (3), was obtained as a colorless oil, which has a molecular formula of C30H46O3 on the basis of HREIMS, showing a molecular ion peak at m/z 454.3454 (calcd for C30H46O3, 454.3447). The IR spectrum suggested the presence of hydroxyl (3428 cm-1), carbonyl (1719 cm-1), and double bond (1657 cm-1) groups. In the 13C NMR (DEPT) spectrum (Table 2), 20 carbon signals including two methyl carbons, nine methylenes, three methines, four quaternary carbons, and two carbons of one double bond, suggested the presence of an ent-kaurene skeleton, which was confirmed by the typical 1H NMR signals (Table 2) of ent-kaurene as follows: δ 4.79 (1H, br. s), 4.73 (1H, br. s), 2.63 (1H, br. s), 1.18 (3H, s), and 0.91 (3H, s). Particularly, the NMR signals of the ent-kaurane moiety were in accordance with those of ent-kaurenoic acid.7 On the other hand, the remaining carbon signals were composed of two olefinic carbons of a trisubstituted double bond, three methyl carbons, one methylene, and four methines (two oxygenated), together with the 1H NMR signals at δ 5.43 (1H, br. s), 5.15 (1H, br. d, J = 8.4 Hz), 4.02 (1H, t, J = 3.3 Hz), 1.81 (3H, s), 0.96, and 0.83 (each 3H, d, J = 6.8 Hz), resembled those of (3R, 4R, 6S)-3, 6-dihydroxymenth-1-ene.15, 16 As shown in Figure 3, the cyclohexene ring in the monoterpene moiety should have half chair configuration. The coupling constant of H-3′ (br. d, J = 8.4 Hz) indicated a trans pseudodiaxial relationship for H-3′ and H-4′ while those of H-6′ (t, J = 3.3 Hz) suggested an equatorial orientation. Consequently, H-3′, H-4′, and H-6′ were determined to be α-, β-, and α-oriented, respectively. The observation of the HMBC correlation (Figure 1) from H-3′ to C-19 as well as the downfield chemical shift of H-3′ at δH 5.15 and the upfield chemical shift of C-19 at δC 177.4, indicated the linkage between the two moieties at C-3′ and C-19 via an ester connection. Therefore, the structure of wedelobatin A was elucidated, as shown in Figure 1.
NMR data of compounds 3 and 4 (13C NMR, 100 MHz, 1H NMR, 500 MHz; CDCl3)
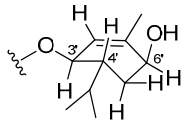
Configuration of the monoterpene moiety
Wedelobatin B (4), was obtained as a colorless oil, and determined molecular formula of C30H44O3 on the basis of HREIMS, showing a molecular ion peak at m/z 452.3283 (calcd for C30H44O3, 452.3290). The IR spectrum suggested the presence of hydroxyl (3429 cm-1), carbonyl (1716 cm-1), and double bond (1654 cm-1) groups. The 1H and 13C NMR data (Table 2) of 4 were similar to those of 3. The major difference found was the presence of one more olefinic proton at δH 5.23 (dd, J = 3.5, 2.7 Hz) and two olefinic carbons of a trisubstituted double bond in 4. The double bond was located at C-9 based on the HMBC correlations from H-11 to C-8, C-10, and C-13, which was confirmed by the fact that the NMR data of the ent-kaurene moiety were consistent with those of 5. Hence, the structure of wedelobatin B was determined, as shown in Figure 1.
Experimental Section
General Experimental Procedures
Optical rotations were measured on a Jasco P-1020 polarimeter. IR spectra were obtained by using a Bruker Tensor 27 FT-IR spectrometer with KBr pellets. NMR spectra were acquired with a Bruker AVANCE Ⅲ-600, Bruker DRX-500 or Bruker AV-400 instrument at room temperature. ESIMS and HREIMS were recorded on a Bruker HCT/Esquire and Waters AutoSpecP776 mass spectrometers. Silica gel (200-300 mesh, Qingdao Marine Chemical Co., Ltd., China), MCI gel CHP-20P (75-150 μm, Mitsubishi Chemical Corporation, Japan), Sephadex LH-20 (Amersham Biosciences, Sweden) and Chromatorex C-18 (40-75 μm, Fuji Silysia Chemical Ltd., Japan) were used for normal pressure column chromatography. MPLC was performed on a Büchi Sepacore System including pump manager C-615, pump modules C-605, and fraction collector C-660 (Büchi Labortechnik AG, Switzerland) and columns were packed with Chromatorex C-18 (40-75 μm, Fuji Silysia Chemical Ltd., Japan). Preparative HPLC was performed on an Agilent 1200 liquid chromatography apparatus with a Zorbax SB-C18 column (5 μm, 9.4 mm × 150 mm). Fractions were monitored and analyzed by TLC (Qingdao Marine Chemical Co., Ltd., China) and spots were visualized by heating silica gel plates immersed in vanillin-H2SO4 in EtOH.
Plant Material
The whole plants of W. trilobata were collected in Pu'er City of Yunnan Province, China, in November 2010, and identified by Mr. Yu Chen of Kunming Institute of Botany, Chinese Academy of Sciences. The voucher specimen (BBP0311) was deposited at BioBioPha Co., Ltd.
Extraction and Isolation
The air-dried, powdered whole plants of W. trilobata (10.0 kg) were extracted with 95% ethanol at room temperature. The alcohol extract was concentrated to derive a residue (1180 g), which was fractionalized by silica gel column chromatography eluted with a solvent system of petroleum ether (PE)-acetone and then MeOH to yield fractions 1-6. Fraction 1 (30 g), eluted with 10% acetone, was further isolated and purified by recrystallization from PE-acetone to afford 5 (17.5 mg). Fraction 2 (48 g), eluted with 15% acetone, was further separated by silica gel column (CHCl3-acetone, 60:1), and then by preparative HPLC (CH3CN-H2O, 50%→70%, 10 mL/min) to derive 3 (4 mg), 4 (4 mg), 12 (48 mg), and 13 (8 mg). Fraction 3 (28 g), was eluted using 20% acetone, and subsequently subjected to a silica gel column with a gradient elution (PE-acetone, 40:1→15:1) to yield fractions 3a-3c. Fraction 3a (6.5 g) yielded 6 (40 mg) and 8 (166 mg) after passing over a MCI gel (MeOH, 100%) and MPLC (MeOH-H2O, 75%→82%, 10 mL/min). 1 (24 mg) and 11 (70 mg) were purified from fraction 3b (7.8 g) through a silica gel column (PE-acetone, 8:1). Fraction 3c (8.5 g) was further separated by silica gel (PE-acetone, 8:1), RP-18 (MeOH-H2O, 70%), and silica gel-AgNO3 (PE-acetone, 3:1→2:1) to yield 2 (16 mg), 7 (170 mg), 9 (25 mg), and 10 (9 mg). Fraction 5 (4.2 g), was eluted using 30% acetone, and purified using a silica gel column (PE-EtOAc, 4:1→3:1) to afford 14 (8 mg) and 15 (22 mg).
3α-Tigloyloxypterokaurene L3 (1)
amorphous powder; [α]D19 -72.9 (c 0.24, CHCl3); IR (KBr) νmax 3513, 2943, 2933, 2859, 1701, 1652, 1450, 1380, 1274, 1258, 1209, 1170, 1152, 1133, 1041, 970 cm-1; 1H NMR and 13C NMR data, see Table 1; ESIMS (pos.) m/z 439 [M + Na]+; HREIMS m/z 416.2554 (calcd for C25H36O5, 416.2563).
ent-17-Hydroxykaura-9(11), 15-dien-19-oic acid (2)
amorphous powder; [α]D19-43.6 (c 0.25, CHCl3); IR (KBr) νmax 3427, 3031, 2928, 2969, 1693, 1639, 1464, 1377, 1228, 1158, 1118, 1003, 988 cm-1; 1H NMR and 13C NMR data, see Table 1; ESIMS (neg.) m/z 315 [M-H]-, HREIMS m/z 316.2028 (calcd for C20H28O3, 316.2038).
Wedelobatin A (3)
oil; [α]D22-121.8 (c 0.41, CHCl3); IR (KBr) νmax 3428, 2956, 2929, 2872, 2853, 1719, 1657, 1465, 1447, 1386, 1369, 1227, 1149, 1015 cm-1; 1H NMR and 13C NMR data, see Table 2; ESIMS (pos.) m/z 477 [M + Na]+, HREIMS m/z 454.3454 (calcd for C30H46O3, 454.3447).
Wedelobatin B (4)
oil; [α]D21-29.2 (c 0.31, CHCl3); IR (KBr) νmax 3429, 2957, 2929, 2870, 1716, 1654, 1464, 1377, 1219, 1144, 1045, 1015, 985 cm-1; 1H NMR and 13C NMR data, see Table 2; ESIMS (pos.) m/z 475 [M + Na]+, HREIMS m/z 452.3283 (calcd for C30H44O3, 452.3290).
Electronic Supplementary Material
Supplementary material is available in the online version of this article at http://dx.doi.org/10.1007/s13659-013-0029-4 and is accessible for authorized users.
Notes
Acknowledgements
The authors acknowledge the National Basic Research Program of China (973 Program, 2009CB522300), and the "West Light" program of Chinese Academy of Sciences.
Compliance with Ethical Standards
References
-
1.Q. T. That, J. Jossang, A. Jossang, P. P. N. Kim, G. Jaureguiberry, J. Org. Chem. 72, 7102-7105 (2007) CrossRef PubMed Google Scholar
-
2.N. Balekar, T. Nakpheng, N. G. Katkam, T. Srichana, Phytomedicine 19, 1178-1184 (2012) CrossRef PubMed Google Scholar
-
3.F. Bohlmann, J. Ziesche, R. M. King, H. Robinson, Phytochemistry 20, 751-756 (1981) CrossRef PubMed Google Scholar
-
4.H. S. Vieira, J. A. Takahashi, M. A. D. Boaventura, Fitoterapia 72, 854-856 (2001) CrossRef PubMed Google Scholar
-
5.Y. Qiang, D. L. Du, Y. J. Chen, K. Gao, Helv. Chim. Acta 94, 817-823 (2011) CrossRef PubMed Google Scholar
-
6.W. F. Roynolds, R. G. Enríquez, L. I. Escobar, X. Lozoya, Can. J. Chem. 62, 2421-2425 (1984) CrossRef PubMed Google Scholar
-
7.M. Hutchison, P. Lewer, J. MacMillan, J. Chem. Soc., Perkin Trans. 1 10, 2363-2366 (1984) PubMed Google Scholar
-
8.B. D. Morris, L. D. Charlet, S. P. Foster, J. Chem. Ecol. 35, 50-57 (2009) CrossRef PubMed Google Scholar
-
9.H. A. Jung, E. J. Lee, J. S. Kim, S. S. Kang, J. H. Lee, B. S. Min, J. S. Choi, Arch. Pharm. Res. 32, 1399-1408 (2009) CrossRef PubMed Google Scholar
-
10.C. Y. Ragasa, W. G. Padolina, B. F. Bowden, S. X. Li, D. M. Tapiolas, J. C. Coll, J. Nat. Prod. 56, 386-393 (1993) CrossRef PubMed Google Scholar
-
11.M. Ahmed, J. Jakupovic, V. Castro, Phytochemistry 30, 1712-1714 (1991) CrossRef PubMed Google Scholar
-
12.E. A. Silva, J. A. Takahashi, M. A. D. Boaventura, A. B. Oliveira, Phytochemistry 52, 397-400 (1999) CrossRef PubMed Google Scholar
-
13.C. Li, D. Lee, T. N. Graf, S. S. Phifer, Y. Nakanishi, S. Riswan, F. M. Setyowati, A. M. Saribi, D. D. Soejarto, N. R. Farnsworth, J. O. Falkinham Ⅲ, D. J. Kroll, A. D. Kinghorn, M. C. Wani, N. H. Oberlies, J. Nat. Prod. 72, 1949-1953 (2009) CrossRef PubMed Google Scholar
-
14.H. S. Santos, F. W. A. Barros, M. R. J. R. Albuquerque, P. N. Bandeira, C. Pessoa, R. Braz-Filho, F. J. Q. Monte, J. H. Leal-Cardoso, T. L. G. Lemos, J. Nat. Prod. 72, 1884-1887 (2009) CrossRef PubMed Google Scholar
-
15.M. D. R. Cuenca, C. A. N. Catalan, J. G. Díaz, W. Herz, J. Nat. Prod. 54, 1162-1164 (1991) CrossRef PubMed Google Scholar
-
16.M. L. Wu, D. Z. Zhang, Q. J. Xu, R. R. Xie, Q. Q. Li, Zhongcaoyao 41, 681-685 (2010) PubMed Google Scholar
Copyright information
© The Author(s) 2013
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
