


New apotirucallane-type triterpenoids from Chisocheton paniculatus
Abstract
Two new apotirucallane triterpenoids, namely chisiamols G(1) and H(2), featuring a 21, 23-lactone, together with five known triterpenoids, were isolated from the twigs of Chisocheton paniculatus. The structures of the new compounds were elucidated on the basis of spectroscopic and chemical methods.Keywords
Chisocheton paniculatus apotirucallane triterpenoids chisiamols G and HIntroduction
The genus Chisocheton (Meliaceae) comprising about 50 species is mainly distributed in India and Malaysia, which is traditionally used to treat stomach and kidney problems, backache, fever, rheumatism and malaria. Chisocheton paniculatus is the only species growing in southern China.1 Phytochemical studies of genus Chisocheton have led to the isolation of sesquiterpenoids, triterpenoids and limonoids, some of which showed anti-inflammatory, anti-feedant, anti-fungal and cytotoxic activities. Previous phytochemical investigation on C. paniculatus resulted mainly in the isolation of apotirucallane-type triterpenoids.2 In the present study, two new apotirucallane triterpenoids, named chisiamols G(1) and H(2), together with five known triterpenoids, chisiamol A(3), 2m sapelin B(4), 3 3α-acetoxy-21, 24R-epoxyapotirucall-14-ene-7α, 23R, 25-triol (5), 2m chisiamol B(6), 2m and 3α-acetoxy-21, 23-epoxyapotirucall-14-ene-7α, 21R, 24, 25-tetrol(7)2m were isolated from the leaves and stems of C. paniculatus (Figure 1). Herein, we describe the isolation and structural elucidation of these new compounds.
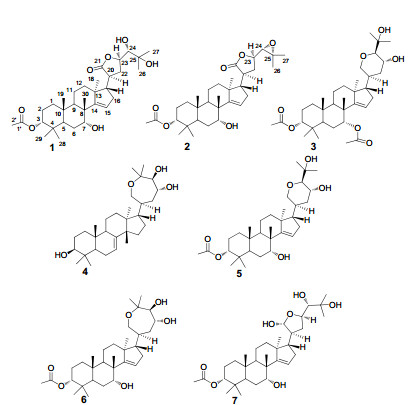
Chemical structures of compounds 1-7
Results and Discussion
Compound 1 was obtained as a white amorphous powder. Its molecular formula was determined to be C32H50O7 by HRESIMS m/z [M + Na]+ 569.3458 (calcd for C32H50O7Na, 569.3454). The IR absorptions at 3448, 1720 and 1630 cm-1 indicated the presence of hydroxyl, ester carbonyl, and double bond groups, respectively. The 1H NMR spectrum showed seven tertiary methyls at δH 0.85, 0.93, 0.96, 1.09, 1.12, 1.24, and 1.28 (each 3H, s), one acetyl methyl at δH 2.06 (3H, s), one olefinic proton at δH 5.45, and four proton signals at δH 3.26 (d), 3.95 (br. s), 4.66-4.74 (m), and 4.63 (br. s) attributable to the protons of oxygenated methines (Table 1). All the 32 carbon resonances were resolved in the 13C NMR spectrum, and further classified by DEPT and HSQC experiments as eight methyls, seven sp3 methylenes, eight sp3 methines (four oxygenated), five sp3 quaternary carbons (one oxygenated), two ester carbonyls (δC 173.2 and 181.6), and one trisubstituted double bond (δC 120.8 and 162.2) (Table 1). Careful analysis of its 1H and 13C NMR data indicated that the NMR data of 1 highly resembled those of the coexisting compound 3α-acetoxy-21, 23-epoxyapotirucall-14-ene-7α, 21R, 24, 25-tetrol(7)2m except for the absence of both the proton and carbon signals of the hemiacetal group (δC 96.5; δH 5.29), and the presence of an additional ester carbonyl, suggesting that C-21 hemiacetal in 7 was oxygenated to a lactone group (δC 181.6) in 1. To prove this speculation, an HMBC experiment was performed, in which, the ester carbonyl was placed at C-21 by the correlations from H2-22 (δH 2.18- 2.33) and H-20 (δH 2.81-2.90) to C-21 (δC 181.6). This was supported by 1H-1H COSY spectrum, in which, a proton bearing structural fragment from C-17 to C-24 as depicted in bold bond (Figure 2) was revealed. In addition, the key HMBC correlations from H-3 (δH 4.63) to C-1′ (δC 173.2) assigned an acetoxy group at C-3; the HMBC correlations of Me-30 (δH 1.12)/C-7 (δC 74.4), and H-7 (δH 3.95)/C-6 located a hydroxyl group at C-7 (Figure 2).
1H (400 MHz) and 13C (100 MHz) NMR data of 1 and 2 (CD3OD)
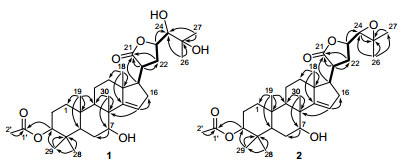
1H-1H COSY (bold lines) and key HMBC (H→C) correlations of 1 and 2
In the ROESY spectrum of 1, the correlations of Me-19/H-3, Me-29/H-3, Me-19/Me-30, and Me-30/H-7, indicated that Me-19, Me-29, Me-30, H-3, and H-7 were cofacial and were randomly assigned to be β-oriented. Subsequently, the OESY correlations of H-20/H-23, and Me-18/H-20 indicated that they were α-oriented (Figure 4). The small coupling constant (1.9 Hz) between H-23 and H-24 indicated that they were in a gauche relationship, showing that H-24 was β-oriented.4
The structure of compound 1 was further verified by chemical correlation with the coexisting known compound 7. Both compounds 1 and 7 were treated with LiAlH4 to afford a common compound 8 (Figure 3), 5 confirming the structural assignment for compound 1. The structure of 8 was assigned as the reduced derivative of compound 7 based on the spectroscopic data. In the ESIMS, the [M + Na]+ peak at m/z 531.4, [2M + Na]+ peak at m/z 1039.7 and [M + HCOO]- peak at m/z 553.7 were all consistent with the molecular formula C30H52O6 of 8 with six hydroxyl groups. A comparison of 1H NMR spectra of 7 and 8 was particularly informative. The absence of the acetyl methyl (δH 2.06 in 7) and the upfield shifted H-3 at δH 3.33 (δH 4.63 in 7) of 8 indicated the presence of OH-3 in 8. The remaining two hydroxyls in 8 were assigned to C-21 and C-23 by the chemical shifts of H2-21 at δH 3.45 and δH 3.77 (each 1H, J = 11.0 Hz) and H-23 at δH 4.08, respectively.
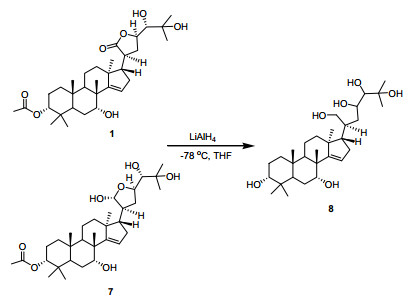
Reduction of compounds 1 and 7 to afford 8
Hence, the structure of 1, namely chisiamol G, was assigned as 3α-acetoxy-7α, 24, 25-trihydroxyl-21, 23-epoxyapotirucall-14-ene-21-one, an apotirucallane-type triterpenoid possessing a five-membered lactone formed between C-21 and C-23.
Compound 2 was obtained as a white amorphous powder. Its molecular formula C32H48O6 was assigned by HRESIMS at m/z [M + Na]+ 551.3348 (calcd for C32H48O6Na, 551.3349). Its IR spectrum exhibited absorption bands for hydroxyl (3432 cm-1), ester carbonyl (1724 cm-1), and double bond (1630 cm-1) functionalities. A comparison of the 13C NMR spectra (Table 1) of compounds 1 and 2 revealed that their structures were close related, except for that two oxygenated carbon signals at δC 79.5 (C-24) and at δC 73.9 (C-25) in 1 were significantly upfield shifted to δC 66.4 (C-24) and δC 59.4 (C-25) in 2, respectively, suggesting that an 24, 25-epoxide ring was present in 2 instead of the 24, 25-diol motif in 1. This was consistent with the fact that the molecular weight of 2 was 18 mass units less than that of 1, and was further supported by the almost identical chemical shifts of C-24 and C-25 of 2 as compared with those of 7α-hydroxyl-21α-acetoxy-21, 23-epoxyapotirucall-24, 25-epoxide-14-ene-3-one and 3α, 7α-di-hydroxyl-21α-acetoxy-21, 23-epoxyapotirucall-24, 25-epoxide-14-ene, both of which possessing an 24, 25-epoxide were respectively reported as compounds A and D in the literature.2a The structure of 2 was finally confirmed by 2D NMR spectra, especially HMBC (Figure 2) and ROESY (Figure 4) spectra. In the ROESY experiment, the correlations of Me-29/H-3, Me-19/Me-30, and Me-30/H-7, indicated that Me-19, Me-29, Me-30, H-3, and H-7 were cofacial and were randomly assigned to be β-oriented. Furthermore, the ROESY correlations of H-20/H-23, and Me-18/H-20 indicated that Me-18, H-20, and H-23 were cofacial and α-oriented. The 24, 25-epoxide ring was assigned to be α-directed by the ROESY correlations of H-22α/H-23 and H-22β/H-24 (Figure 4).6 The chemical shifts of C-24 (δC 66.4) and C-25 (δC 59.4), and the coupling constant (J23, 24 = 8.1 Hz) between H-23α and H-24 further supported the α-orientation of the epoxide in 2 as compared with the corresponding data of the bruceajavanones A-E.6 The structure of 2 was thus established as 3α-acetoxy-7α-hydroxyl-21, 23-epoxyapotirucall-24, 25-epoxide-14-ene-21-one, namely chisiamol H.
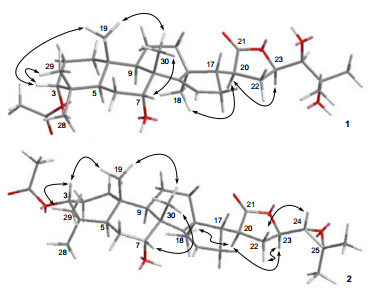
Key NOE (H↔H) correlations of 1 and 2
All isolated compounds(1-7) were tested in an antimicrobial assay against Helicobacter pylori-SS1 in vitro according to standard protocols, in which Minimum inhibitory concentration (MIC) > 100 μg/mL was defined as inactive7, and metronidazole (Shanghai Henshan Pharmaceutical Company, Ltd. with a purity of 99%; MIC = 0.312 μg/mL) was used as the positive control. However, none of these compounds showed activity (MIC > 50 μg/mL).
Experimental Section
General Experimental Procedures. Optical rotations were measured with a Perkin-Elmer 341 polarimeter. UV spectra were measured on a Shimadzu UV-2550 spectrophotometer. IR spectra were recorded on a Perkin-Elmer 577 spectrometer using KBr disks. NMR spectra were measured on a Bruker AM-400 spectrometer. ESIMS was carried out on a Finnigan LCQ-DECA instrument, and HRESIMS spectra were made on a Bruker Daltonics micrOTOFQII or a Waters-Micromass Q-TOF Ultima Global electrospray mass spectrometer. All solvents used were of analytical grade (Shanghai Chemical Reagents Company, Ltd.). Different absorbents were employed for column chromatography: silica gel (200-300 mesh) and silica gel H (Qingdao Haiyang Chemical Co. Ltd.); RP-18 reversed silica gel (150-200 mesh, Merck); MCI gel (CHP20P, 75-150 ím, Mitsubishi Chemical Industries Ltd.); Sephadex LH-20 gel (Amersham Biosciences). Fractions were monitored by TLC (silica gel GF254, 0.25 mm, Merck), and spots were visualized by heating silica gel plates sprayed with 10% H2SO4 in ethanol. Semipreparative HPLC was performed on a Waters 1525 pump equipped with a Waters 2489 detector and an YMC-Pack ODS-A column (10 × 250 mm, S-5 μm, 12 nm).
Plant Materal. The leaves and stems of C. paniculatus, were collected in Mengla County, Yunnan Province of China in September of 2009, and the plant was identified by Professor You-Kai Xu of Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences. A voucher specimen (accession number CS-2009-2Y) has been deposited in the Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
Extraction and Isolation. Dried leaves and stems of C. paniculatus (6 kg) were extracted by 95% EtOH at room temperature. The crude extract (70 g) was dissolved in H2O (500 mL) and then extracted with EtOAc (200 mL × 3). The EtOAc extract (24 g) was subjected to an MCI column (75-150 μm, 8 × 30 cm, MeOH/H2O, 50:50 to 90:10, v/v) to give fractions E-K. Fraction Ⅰ (1.80 g) was chromatographed on a silica gel column eluted with petroleum ether/Me2CO (300-400 mesh, 4 × 20 cm, 10:1 to 4:1) to yield four fractions (Ⅰ1-4). Fraction Ⅰ4 (193 mg) was subjected to a silica gel column (300-400 mesh, 3 × 18 cm, CH2Cl2/MeOH, 150:1, 100:1, 70:1, 50:1, 30:1, 20:1, 10:1) to give four fractions (Ⅰ4a-d). Purification of fraction Ⅰ4b (30 mg) by a RP-18 silica gel column (150-200 mesh, 3 × 15 cm, MeOH/H2O, 70:30 to 80:20) gave 1 (20 mg). Fraction J (5.50 g) was chromatographed on a silica gel column eluted with petroleum ether/Me2CO (300-400 mesh, 8 × 20 cm, 10:1 to 6:1) to yield seven fractions (J1-7). Fraction J3 (221 mg) was submitted to a silica gel column (300-400 mesh, 3 × 20 cm, CH2Cl2/MeOH, 100:1, 80:1, 60:1) to give three fractions (J3a-c). Purification of fraction J3a (12 mg) with Sephadex LH-20 (3 × 100 cm, EtOH) yielded 2 (3 mg). Fraction J3b (73 mg) was applied to a RP-18 silica gel column (150-200 mesh, 3 × 15 cm, MeOH/H2O, 80:20 to 90:10) to give fractions J3b1 and J3b2. Fraction J3b2 (14 mg) was further purified by preparative HPLC (eluting with MeCN/H2O, 85:15 to 95:5, from 0 to 20 min, 3 mL/min) afforded 3 (3 mg). Purification of fraction J3c (15 mg) by preparative HPLC (eluting with MeCN/H2O, 90:10 to 95:5, from 0 to 20 min, 3 mL/min) yielded 4 (4 mg). Fraction J5 (358 mg) and J6 (293 mg) were subjected to a silica gel column (300-400 mesh, 3 × 20 cm, petroleum ether/ EtOAc, 10:1, 8:1, 6:1, 4:1) to give two corresponding fractions J5a-b and J6a-b, respectively. Fraction J5a (140 mg) and J6a (255 mg) were applied to a silica gel column (300-400 mesh, 3 × 18 cm, CH2Cl2/MeOH, 100:1, 80:1, 60:1, 40:1) to give three corresponding fractions J5a1-3 and J6a1-3, respectively. Purification of fractions J5a2 (40 mg) and J6a3 (83 mg) were subjected to RP-18 silica gel column (150-200 mesh, 3 × 15 cm, MeOH/H2O, 90:10 to 100:0) to afford 5 (35 mg) and 6 (9 mg). Fraction E (1.34 g) was chromatographed on a silica gel column eluted with petroleum ether/Me2CO (300-400 mesh, 6 × 20 cm, 10:1 to 1:1) to yield seven fractions (E1-7). Purification of fraction E3 (40 mg) was applied to RP-18 silica gel column (150-200 mesh, 3 × 15 cm, MeOH/H2O, 80:20 to 90:10) to afford 7 (20 mg).
Chisiamol G(1): White amorphous powder; [α]D23 - 105 (c = 0.27, MeOH); UV (MeOH) λmax (log ε) 202.4 (4.3) nm; IR (KBr) νmax 3448, 2939, 2871, 1765, 1720, 1630, 1458, 1385, 1169, 1034 cm-1; 1H NMR and 13C NMR see Table 1; ESIMS m/z [M + Na]+ 569.4; HRESIMS m/z [M + Na]+ 569.3458 (calcd for C32H50O7Na, 569.3454).
Chisiamol H(2): White amorphous powder; [α]D23 - 69 (c = 0.30, MeOH); UV (MeOH) λmax (log ε) 201.6 (4.15) nm; IR (KBr) νmax 3432, 2937, 2871, 1774, 1724, 1630, 1458, 1385, 1250, 1188, 1024, 818 cm-1; 1H NMR and 13C NMR see Table 1; ESIMS m/z [M + Na]+ 551.4; HRESIMS m/z [M + Na]+ 551.3348 (calcd for C32H48O6Na, 551.3349).
Reduction of Compounds 1 and 7 with LiAlH4. To a cooled solution (- 78 ℃) of compound 1 or 7 (5.0 mg) in dry THF (2 mL), LiAlH4 (3 mg) was added under nitrogen atmosphere. After stirring at the same temperature for 2 h, the reaction mixture was then allowed to r.t. and kept stirring overnight. After workup, the reaction mixture was diluted with H2O (2 mL) and then extracted with EtOAc (3 × 5 mL). The combined organic phase were dried with anhydrous Na2SO4 and concentrated in vacuo to obtain the crude product, which was purified by a silica gel column eluted with CH2Cl2/MeOH (300-400 mesh, 3 × 12 cm, 50:1 to 10:1) to give compound 8 (about 3.0 mg).
Compound 8: ESIMS m/z 531.4 [M + Na]+, m/z 1039.7 [2M + Na]+, m/z 553.7 [M + HCOO]-; 1H NMR (400 MHz, δH ppm, CD3OD): 5.44 (H-15, d, J = 2.6 Hz), 4.08 (H-23, t, J = 6.7), 3.88 (H-17, t-like), 3.45, 3.77 (H-21, ABq, J = 11.0 Hz), 3.33 (H-3, t-like), 3.12 (H-24, d, 1.5 Hz).
Notes
Electronic Supplementary Material
Supplementary material is available in the online version of this article at http://dx.doi.org/10.1007/s13659-012-0065-5 and is accessible for authorized users.
Acknowledgments
Financial support of the National Natural Science Foundation (Grant No. 21072203) and National Science and Technology Major Project "Key New Drug Creation and Manufacturing Program" (No. 2011ZX09307-002-03) of China is gratefully acknowledged. We thank Prof. You-Kai Xu for the identification of the plant material.
References
-
1.(a) Chen, S. K. ; Li, H. ; Chen, P. Y. Flora of China; Science Press: Beijing, 1997; Vol. 43, p 98-99. (b) Mulholland, D. A. ; Parel, B. ; Coombes, P. H. Curr. Org. Chem. 2000, 4, 1011-1054. PubMed Google Scholar
-
2.(a) Connolly, J. D. ; Labbe, C. ; Rycroft, D. S. ; Taylor, D. A. H. J. Chem. Soc., Perkin Trans. I 1979, 2959-2964. (b) Chatterjee, A. ; Nayak, L. ; Das, B. ; Patra, A. ; Dhara, K. P. ; Mukherjee, K. ; Banerji, J. Indian J. Chem., Sect. B 1989, 28, 231-236. (c) Bordoloi, M. ; Saikia, B. ; Mathur, R. K. ; Goswami, B. N. Phytochemistry 1993, 34, 583-584. (d) Yadav, R. D. ; Kataky, J. C. S. ; Mathur, R. K. Indian J. Chem., Sect. B 1999, 38, 243-245. (e) Sarmah, G. K. ; Bhattacharyya, N. K. ; Goswami, B. N. ; Barua, P. ; Kataky, J. C. S. J. Indian Chem. Soc. 2003, 80, 1163-1168. (f) Awang, K. ; Lim, C. S. ; Mohamad, K. ; Morita, H. ; Hirasawa, Y. ; Takeya, K. ; Thoison, O. ; Hadi, A. H. Bioorg. Med. Chem. 2007, 15, 5997-6002. (g) Maneerat, W. ; Laphookhieo, S. ; Koysomboon, S. ; Chantrapromma, K. Phytomedicine 2008, 15, 1130-1134. (h) Phongmaykin, J. ; Kumamoto, T. ; Ishikawa, T. ; Suttisri, R. ; Saifah, E. Arch. Pharm. Res. 2008, 31, 21-27. (i) Laphookhieo, S. ; Maneerat, W. ; Koysomboon, S. ; Kiattansakul, R. ; Chantrapromma, K. ; Syers, J. K. Can. J. Chem. 2008, 86, 205-208. (j) Mohamad, K. ; Hirasawa, Y. ; Litaudon, M. ; Awang, K. ; Hadi, A. H. ; Takeya, K. ; Ekasari, W. ; Widyawaruyanti, A. ; Zaini, N. C. ; Morita, H. Bioorg. Med. Chem. 2009, 17, 727-730. (k) Xie, B. J. ; Yang, S. P. ; Zhang, C. ; Yue, J. M. Chin. J. Chem. 2009, 27, 1805-1810. (l) Yang, M. H. ; Wang, J. S. ; Luo, J. G. ; Wang, X. B. ; Kong, L. Y. J. Nat. Prod. 2009, 72, 2014-2018. (m) Yang, M. H. ; Wang, J. S. ; Luo, J. G. ; Wang, X. B. ; Kong, L. Y. Can. J. Chem. 2012, 90, 199-204. PubMed Google Scholar
-
3.S. D. Jolad, J. J. Hoffmann, J. R. Cole, J. Org. Chem. 45, 3132-3135 (1980) CrossRef PubMed Google Scholar
-
4.B. J. Xie, S. P. Yang, H. D. Chen, J. M. Yue, J. Nat. Prod. 70, 1532-1535 (2007) CrossRef PubMed Google Scholar
-
5.W. W. Zi, S. Y. Yu, D. W. Ma, Angew. Chem. Int. Ed. 49, 5887-5890 (2010) CrossRef PubMed Google Scholar
-
6.L. Pan, Y. W. Chin, H. B. Chai, T. N. Ninh, D. D. Soejarto, A. D. Kinghorn, Bioorg. Med. Chem. 17, 2219-2224 (2009) CrossRef PubMed Google Scholar
-
7.D. H. Kwon, M. Kato, F. A. K. El-Zaatari, M. S. Osato, D. Y. Graham, FEMS Microbiol. Lett. 188, 197-202 (2000) CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2012
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
