


Two new neolignans from Manglietia insignis
Abstract
Two new neolignans, manneoinsigins A(1) and B(2), together with four known lignans, were isolated from the leaves and stems of Manglietia insignis. The new compounds were established on the basis of extensive spectroscopic analyses. All compounds except 2 were tested for their cytotoxic activity. Compound 3 showed weak cytotoxic activity against the HL-60 human tumor cell line with the IC50 value of 23.5 μM.Keywords
Magnoliaceae Manglietia insignis neolignan cytotoxic activityIntroduction
Plants of the family Magnoliaceae are mainly distributed in southeastern Asia, which contain more than 250 species and are a rich source of lignans possessing various pharmacological functions.1-9 Structural and biological diversity of lignans in magnoliaceae family prompted us to investigate Magnolia insignis Rehd. et Wils, a plant widely distributed in the west of China and being partly used as a substitute of M. officinalis in Yunnan and Sichuan provinces of China.4,10 As a result, two new neolignans, manneoinsigins A (1) and B (2), together with other four known lignans, scaphopetalone (3),11 mesosecoisolariciresinol (4),12 lariciresinol (5)13 and evafolin B (6),14 were isolated. Herein, the isolation, structural elucidation, and their cytotoxic activity are described.
Results and Discussion
Manneoinsigin A (1) was obtained as yellow gum. The negative ESIMS of 1 showed characteristic peaks for [M – H]– and [M – H + 2]– with the ratio being 3:1, which suggested the presence of a chlorine atom. The molecular formula of 1 was determined as C18H19O4Cl by its HRESIMS with a pseudomolecular ion peak at m/z 333.0897 [M – H]–, corresponding to nine unsaturation degrees. The 1H NMR spectrum displayed signals due to two sets of ABX-type aromatic systems (δH 7.28, 6.91 and 7.24; δH 7.08, 6.84 and 7.02), one terminal double bond (δH 5.00, 5.05 and 5.97), two methylenes (δH 3.33 and 3.61), and two methines (δH 4.67 and 3.85) (Table 1). The 13C NMR (DEPT) spectrum exhibited eighteen carbon signals, consisting of fourteen sp2 ones (two of which were signals of allyl group at δC 115.6 and 139.4), two methylenes, and two oxygenated methines (Table 1). Comparison of 1D NMR spectra of 1 with those of magnolol showed that they possessed the same skeleton.15 The differences could be rationalized to be that the allyl group in magnolol was replaced by a 1, 2-dihydroxy-3-chlorine-propyl group in 1. This was further supported by the HMBC correlations of H-7 (δH 4.67, 1H, d, J = 5.9 Hz) with C-1 (δC 134.2), C-2 (δC 131.3), C-6 (δC 128.2), C-8 (δC 76.9) and C-9 (δC 47.3), together with the 1H-1H COSY correlations of H-7/H-8/H2-9 (Fig. 2). Furthermore, the HMBC correlations of H-7 with C-1, C-2 and C-6 indicated that the 1, 2-dihydroxy-3-chlorine-propyl group was located at C-1 (Fig. 2). The chlorine atom was deduced to be located at C-9 by the characteristic chemical shift of C-9 at δC 47.3.
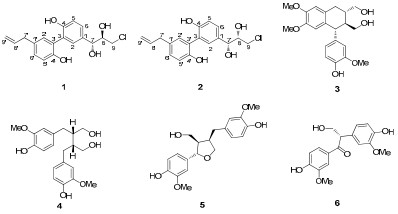
The structures of compounds 1–6
1H and 13C NMR spectroscopic assignments of compounds 1 and 2a
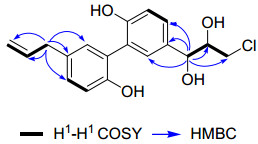
Selected 2D NMR correlations of 1
Manneoinsigin B (2) was obtained as yellow gum. The presence of a chlorine atom was deduced by the characteristic peaks for [M – H]– and [M – H + 2]– with the ratio being 3:1 in its negative ESIMS. The molecular formula of 2 was deduced as C18H19O4Cl from its HREIMS (m/z 334.0964 [M]+). Detailed analysis of the 1D NMR data of 1 and 2 suggested that both of them had the same plane structure (Table 1). It was found that the differences of the 1H NMR chemical shifts of H-7 and H-9 from δH 4.67, 3.61, and 3.33 in 1 changing to δH 4.56, 3.77, and 3.67 in 2 and the 13C NMR chemical shifts of C-7, C-8 and C-9 from δC 75.4, 76.9, and 47.3 in 1 changing to δC 75.8, 76.5, and 48.1. The reason for the minor distinct NMR data of 1 and 2 could be rationalized to the dissimilar configurations of the stereogenic carbons of the side chains.
The OH configurations of two stereogenic centers in 1 and 2 were determined by comparison with the 13C NMR data of the structural similar compounds, erythro-and threo-honokitriol16 and erythro-and threo-1-C-syringylglycerol.17 The difference of 13C NMR chemical shifts of C-7 (δC 76.0) and C-8 (δC 76.2) in erythro-honokitriol is △δC-8–C-7 0.2, which is smaller than that in threo-honokitriol (△δC-8(77.8)–C-7(74.6) 3.2).16 In addition, the difference of 13C NMR chemical shifts of C-7 (δC 74.1) and C-8 (δC 75.3) in erythro-1-C-syringylglycerol is △δC-8–C-7 1.2, which is also smaller than that in its threo-isomer (△δC-8(75.8)–C-7(72.9) 2.9).17 Therefore, the relative configurations of 1 and 2 could also be determined base on the rules as mentioned in the above cases.16,17 In compound 2, the difference of the carbon signals of C-7 (δC 75.8) and C-8 (δC 76.5) is △δC-8–C-7 0.7, which is smaller than that in 1 (△δC-8(76.9)–C-7(75.4) 1.5). Thus, the structures of 1 and 2 were determined as threo-and erythro-configurations, respectively. As for the absolute configuration of C-7 and C-8, there should be four possibilities, i.e. (7R, 8R), (7R, 8S), (7S, 8R), and (7S, 8S). In the literature, four stereoisomers of 1-phenylglycidol with (1R, 2R), (1R, 2S), (1S, 2R), (1S, 2S) configurations were synthesized by an asymmetric method, and the optical data were [α]D18 – 45.5 (c 3.3), [α]D25 – 38.5 (c 2.2), [α]D20 + 37.2 (c 3.4), [α]D21 + 45.0 (c 2.4), respectively.18 By comparison of their optical data with those of threo isomer (1, [α]D23.5 – 11.0) and erythro isomer (2, [α]D20 – 14.6), it was confirmed that the configuration of C-7 and C-8 in 1 and 2 were (7R, 8R) and (7R, 8S), respectively.
Some chemical constituents from the Magnoliaceae family were reported to have obvious cytotoxic activities.1,9 Therefore, all compounds except compound 2 were assayed for their cytotoxicity against the HL-60, SMMC-7721, A-549, MCF-7, and SW-480 human tumor cell lines by the MTT method with cis-platin as positive control.19 Compound 3 showed weak cytotoxic activity against the HL-60 human tumor cell lines with the IC50 values of 23.5 μM. The other compounds did not show cytotoxic activity (IC50 > 40 μM).
Experimental Section
General Experimental Procedures. Optical rotations were measured with a JASCO DIP-370 digital polarimeter. UV spectra were obtained using a Shimadzu UV-2401A spectrophotometer. A BioRad FtS-135 spectrophotometer was used for scanning IR spectroscopy with KBr pellets. 1D and 2D NMR spectra were recorded on Bruker AM-400, DRX-500 and Bruker Avance III-600MHz spectrometers. Unless otherwise specified, chemical shifts (δ) were expressed in ppm with reference to the solvent signals. High resolution electrospray ionization (HRESIMS) were performed on a VG Autospec-3000 spectrometer under 70 eV. Column chromatography was performed using silica gel (200–300 mesh, Qingdao Marine Chemical, Inc., Qingdao, China). Semi-preparative HPLC was performed on an Agilent 1100 liquid chromatograph with a Zorbax SB-C18, 9.4 mm × 25 cm, column. Preparative HPLC was performed on a Shimadzu LC-8A preparative liquid chromatograph with a Shimadzu PRC-ODS (K) column. Fractions were monitored by TLC and spots were visualized by heating the silica gel plates sprayed with 10% H2SO4 in EtOH.
Plant Materal. The leaves and stems of M. insignis (Wall.) Bl. was collected in Kunming Botanic Garden, Yunnan Province, China, in August 2007. The specimen was identified by Prof. Xun GONG and a voucher specimen (No. KIB 2007-08-11) has been deposited at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.
Extraction and Isolation. The air-dried plant material of M. insignis (6.5 kg) was ground and exhaustively percolated three times with 70% aqueous Me2CO at room temperature. The solvent was evaporated in vacuo, and the combined crude extract was dissolved in H2O and partitioned with EtOAc. The EtOAc portion (156 g) was subsequently chromatographed on a silica gel column eluting with CHCl3-Me2CO (1:0, 9:1, 8:2, 2:1, 1:1, and 0:1) and was combined into six groups (A–F) according to TCL analysis. Fraction B (21g) was chromatographed on MPLC eluting with MeOH/H2O (30:70–100:0 gradient system), to give fraction B1–B5. And then fraction B3 was further applied to silica gel CC, and eluted with petroleum ether-acetone (20:1, 12:1, 8:1 and 3:1) to give five fractions B3-1–B3-5. Compounds 1 (7.0 mg, tR = 29 min) and 2 (2.1 mg, tR = 32 min) were obtained from fraction B3-2 by semi-preparative HPLC (42% aq. acetonitrile, 3 mL/min). Fraction B3-3 was chromatographed on Sephadex LH-20 (MeOH) and then successively chromatographed on silica gel CC (CHCl3-ethyl acetate, 25:1, 16:1, 12:1 and 6:1) to give compound 6 (12 mg). Fraction B4 was further purified on silica gel CC (CHCl3-ethyl acetate, 24:1–8:1) and then chromatographed on Sephadex LH-20 (MeOH) to give six fractions B4-1–B4-6. Fraction B4-2 were further subjected to semi-preparative HPLC (53% aq. MeOH, 3 mL/min) to afford 3 (9.0 mg, tR = 18 min) and 4 (14.6 mg, tR = 24 min). Compound 5 (5.6 mg, tR = 18 min) were purified by semipreparative HPLC (55% aq. MeOH, 3 mL/min) from fraction B4-3.
Manneoinsigin A (1): yellow gum; [α]D23.5 – 11.0 (c 0.23, MeOH); UV (MeOH) λmax (log ε) 289 (3.27), 214 (4.01), 191 (3.76) nm; IR (KBr) νmax 3424, 3081, 2924, 2853, 1639, 1619, 1496, 1423, 1230, 915, 826 cm–1; 1H and 13C NMR data, see Table 1; negative ESIMS m/z 333 [M – H]– and isotopic peak m/z 335 [M – H]–; negative HRESIMS m/z 333.0897 [M – H]–(calcd for C18H18O4Cl, 333.0893) and isotopic peak m/z 335.0908 [M – H]– (calcd for C18H18O4Cl, 335.0864).
Manneoinsigin B (2): yellow gum; [α]D23.5 – 14.6 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 289 (3.49), 212 (4.20), 196 (3.84) nm; IR (KBr) νmax 3426, 3080, 2927, 2856, 1638, 1624, 1496, 1230, 917, 830 cm–1; 1H and 13C NMR data, see Table 1; negative ESIMS m/z 333 [M – H]– and isotopic peak m/z 335 [M – H]–; HREIMS m/z 334.0964 [M]+ (calcd for C18H19O4Cl, 334.0972) and isotopic peak m/z 336.0961 [M]+ (calcd for C18H19O4Cl, 336.0942).
Cytotoxicity Assay. The following human tumor cell lines were used: HL-60, MMC-7721, A549, MCF-7, and SW480. All cells were cultured in RPMI-1640 or DMEM medium (Hyclone, Logan, UT), supplemented with 10% fetal bovine serum (Hyclone) at 37 ℃ in a humidified atmosphere with 5% CO2. Cell viability was assessed by conducting colorimetric measurements of the amount of insoluble formazan formed in living cells based on the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma, St. Louis, MO).19 Briefly, 100 μL of adherent cells was seeded into each well of a 96-well cell culture plate and allowed to adhere for 12 h before drug addition, while suspended cells were seeded just before drug addition, both with an initial density of 1×105 cells/mL in 100 μL of medium. Each cell line was exposed to the test compound at various concentrations in triplicate for 48 h, with cisplatin and paclitaxel (Sigma) as positive controls. After the incubation, MTT (100 μg) was added to each well, and the incubation continued for 4 h at 37 ℃. The cells were lysed with 100 μL of 20% SDS-50% DMF after removal of 100 μL of medium. The optical density of the lysate was measured at 595 nm in a 96-well microtiter plate reader (BioRad 680). The IC50 value of each compound was calculated by Reed and Muench's method.20
Notes
Electronic Supplementary Material
Supplementary material is available in the online version of this article at http://dx.doi.org/10.1007/s13659-012-0063-7 and is accessible for authorized users.
Acknowledgments
This project was supported financially by the NSFC (No. 20802082 and 30830115), the projects from the Chinese Academy of Sciences (KSCX2-EW-Q-10 and KSCX1-YW-R-24), the Major State Basic Research Development Program of China (No. 2009CB522300 and 2009CB940900), and the Natural Science Foundation of Yunnan Province (2012FB178 and 20080A007) and the project of recruited top talent of sciences and technology of Yunnan Province (2009C1120).
References
-
1.U. J. Youn, Q. C. Chen, W. Y. Jin, I. S. Lee, H. J. Kim, J. P. Lee, M. J. Chang, B. S. Min, K. H. Bae, J. Nat. Prod. 70, 1687-1689 (2007) CrossRef PubMed Google Scholar
-
2.K. Watanabe, H. Y. Watanabe, Y. Goto, N. Yamamoto, M. Yoshizaki, Jpn. J. Pharmacol. 25, 605-607 (1975) CrossRef PubMed Google Scholar
-
3.K. Watanabe, H. Watanabe, Y. Goto, M. Yamaguchi, N. Yamamoto, K. Hagino, Planta Med. 49, 103-108 (1983) CrossRef PubMed Google Scholar
-
4.K. Watanabe, Gendai Toyo Igaku 7, 54-59 (1986) PubMed Google Scholar
-
5.C. M. Teng, S. M. Yu, C. C. Chen, Y. L. Huang, T. F. Huang, Life Sci. 47, 1153-1161 (1990) CrossRef PubMed Google Scholar
-
6.E. A. Bae, M. J. Han, D. H. Kim, Planta Med. 65, 442-443 (1999) CrossRef PubMed Google Scholar
-
7.T. Namba, M. Tsunezuka, M. Hattori, Planta Med. 44, 100-106 (1982) CrossRef PubMed Google Scholar
-
8.E. J. Sohn, C. S. Kim, Y. S. Kim, D. H. Jung, D. S. Jang, Y. M. Lee, J. S. Kim, Life Sci. 80, 468-475 (2007) CrossRef PubMed Google Scholar
-
9.Y. Fukuyama, Y. Otoshi, K. Miyoshi, K. Nakamura, M. Kodama, M. Nagasawa, T. Hasegawa, H. Okazaki, M. Sugawara, Tetrahedron 48, 377-392 (1992) CrossRef PubMed Google Scholar
-
10.College, J. N. M. Ed. Chinese Drug Dictionary. Shanghai Science and Technology Publishing Co. 1977, 1628-1630. PubMed Google Scholar
-
11.J. C. Vardamides, A. G. B. Azebaze, A. E. Nkengfack, F. R. Van Heerden, Z. T. Fomum, T. M. Ngando, J. Conrad, B. Vogler, W. Kraus, Phytochemistry 62, 647-650 (2003) CrossRef PubMed Google Scholar
-
12.T. Sugahara, S. Yamauchi, A. Kondo, F. Ohno, S. Tominaga, Y. Nakashima, T. Kishida, K. Akiyama, M. Maruyama, Biosci. Biotechnol. Biochem. 71, 2962-2968 (2007) CrossRef PubMed Google Scholar
-
13.T. Masuda, J. Akiyama, A. Fujimoto, S. Yamauchi, T. Maekawa, Y. Sone, Food Chem. 123, 442-450 (2010) CrossRef PubMed Google Scholar
-
14.X. Luo, S. Wu, Y. Ma, D. Wu, Acta. Bot. Yunnanica 23, 368-372 (2001) PubMed Google Scholar
-
15.X. M. Deng, Y. X. Cheng, J. Zhou, N. H. Tan, Z. T. Ding, Acta Bot. Yunnanica 23, 121-125 (2001) PubMed Google Scholar
-
16.C. C. Shen, C. L. Ni, Y. C. Shen, Y. L. Huang, C. H. Kuo, T. S. Wu, C. C. Chen, J. Nat. Prod. 72, 168-171 (2009) CrossRef PubMed Google Scholar
-
17.H. Otsuka, M. Takeuchi, S. Inoshiri, T. Sato, K. Yamasaki, Phytochemistry 28, 883-886 (1989) CrossRef PubMed Google Scholar
-
18.M. Takeshita, R. Yaguchi, N. Akutsu, Tetrahedron:Asymmetry 3, 1369-1372 (1992) CrossRef PubMed Google Scholar
-
19.Z. R. Ping, Z. Hongjie, L. Zhongwen, Z. Yulin, S. Handong, Phytochemistry 31, 4237-4240 (1992) CrossRef PubMed Google Scholar
-
20.L. J. Reed, H. Muench, In, Am. J. Hyg. 27, 493-497 (1938) PubMed Google Scholar
Copyright information
© The Author(s) 2012
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
