


Exploring of drug leads from diversity-oriented Michael-acceptor library derived from natural products
Abstract
A potential strategy for drug lead identification and in-active natural products re-discovery is elaborated. Starting from fifteen structurally diverse natural products, a focused library featured by Michael acceptors is constructed with IBX mediated oxidation. Biological assay on five tumor cell lines indicates that four Michael acceptors, 8a, 11a, 12a, 14a, are with improved cytotoxicity(3-10 folds more potent than the parent compounds), which merit further investigations. Further thiol-sensitive assay of the active hit 8a revealed that it was an irreversible Michael acceptor. The results suggest that the strategy is not only effective and relatively high discovery rate(28%), but also resource saving.Keywords
drug leads identification in-active natural products re-discovery Michael acceptors anti-tumor activityIntroduction
Drug-lead identification is the first step of drug discovery. A good start may set a success keynote for the whole process. So it is worthwhile to pay more attention on drug lead identification. Though plenty methods, including random screening, high-throughput screening from database, fragment based lead design and computer aided lead identification, are available for drug lead discovery1, 2, some challenges remain un-addressed. The discovery efficiency and structure diversity are often low, and the leads are always hampered on the way to drug candidates. In one word, the lead quality is frequently unsatisfactory3, 4.
On the other hand, the abundant structure and function diversity fueled by natural products has provided a steady flow for the novel drugs. Many drugs used in clinic are natural products or compounds derived from natural products5-7. It is particularly true in anti-cancer and anti-infectious therapy field. We would like to dig out something valuable from this underestimated repository.
What's more, in the drug lead identification process, the major attentions are focused on the active hits, leaving vast amount of counterparts under-valued. And what shocks us most is that plenty of the "in-active" natural products are abundant in plants. So how to make sustainable use of these "in-active" natural products becomes a highly valuable project. We wonder that if we can turn the "trash" to the "treasure" by incorporation of pharmaceutically privileged structure moiety through some simple chemical transformations.
Based on the reasons mentioned above, we would like to combine the diverse natural products and the pharmaceutically privileged moiety together so as to identify novel drug leads. So what moiety will be chosen? The α, β-unsaturated ketone moiety came to our sight and became our right choice.
It is generally believed that Michael acceptors form covalent bond with the active site cysteine of the proteases to solicit a biological effect8-11. The Cysteine proteases, including papains, cathepsins, calpains, caspases and lugumain, represent a biologically important clan of protein, which are important therapeutic target of tumor, inflammation and auto-immune disease12-14. So it is no surprise that Michael acceptors have the potential of anti-tumor activity. In 2005, a Michael acceptor derived from oleanic acid, TP235, was approved as an orphan drug for treatment of pancreatic cancer and also used as an agent for treatment of diabetes associated chronic kidney disease in late clinical trial15, 16. This discovery has provoked the keen interests in Michael acceptors. Since then, more and more successful examples in drug discovery spring up. A cruzain inhibitor, CRA-3386, was approved into clinical trial for treatment of Chagas disease17. Another Michael acceptor, AG7088, entered into clinical trial Ⅱ for therapy of rhinovirus infection18. Schust J. et al. identified a novel Michael acceptor, static, as a selective STAT3 activation inhibitor from a compound library, and proved that the Michael acceptor moiety was its key pharmacophore (Figure 1)19. The pioneer work leads us to an idea that identification of novel drug leads from structurally diverse and functionally under-mined natural products by introduction Michael acceptor moiety.
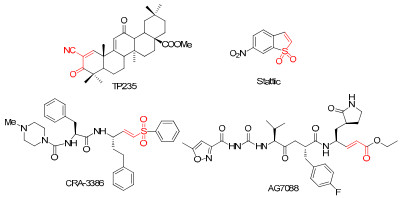
Typical Michael acceptors
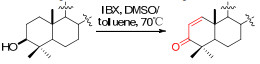
With the idea in mind, we then started the execution of the plan. Though plenty of methods available in our chemical tool box, including Pd(OAc)2 catalyzed Saegusa oxidation20, benzeneselenenylation-oxidative elimination sequence21, both the methods require trivial operations and relative harsh reaction conditions. Fortunately, we have a newly invented method namely IBX mediated oxidation available22, which features with higher efficiency and simpler operations. The power of the method is not fully exemplified, especially in complex natural products.
Herein, we reported the construction of a small diversityoriented combinatorial library derived from natural products, which featured with α, β-unsaturated carbonyl moiety using IBX mediated oxidation.
Results and Discussion
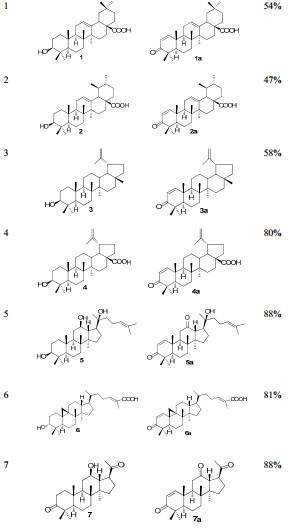
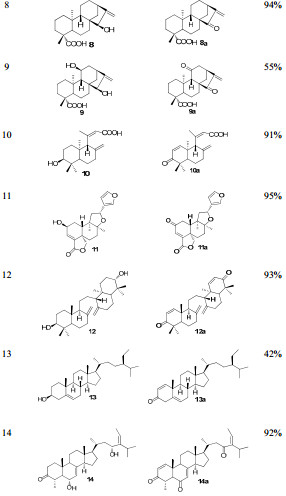
Starting from structurally diverse natural products, we prepared fourteen Michael acceptors efficiently (Table 1).
Preparation of Michael acceptors
All the substrates and the Michael acceptors were evaluated on five human tumor cell lines, including HL-60, SMMC-7721, A-549, SK-BR-3 and PANC-1, using 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) method. And anticancer drug cisplatin (DDP) was used as the positive control (Table 2).
In Vitro anti-tumor assay of the substrates and Michael acceptors
As summarized in Table 2, four Michael acceptors, 8a, 11a and 12a, 14a, are found to be with significantly improved antitumor activity. Especially, compounds 8a and 12a exhibit single digit micro-molar inhibitory activity against HL-60, SK-BR-3 and PANC-1 cell lines, which are up to 10 times more potent than the corresponding parent compounds. It's also pleased to know that the parent compounds 8 and 12 are relatively abundant in plants, which are worthy of our further attentions.
Moreover, we also investigated the possible action mode of the active hit 8a in a thiol-sensitive assay using an NMR method, which was developed by Appendino G. et al23. The method was proved effective for identifying Michael acceptors, and sorting them into reversible and irreversible thiol sinks. Upon treatment of two equivalents of cysteamine, a known biologically relevant model thiol24, in deuterated DMSO, a Michael adduct of 8b was formed instantaneously (Figure 2), and the adduct was irreversible by diluting with CDCl3. The results reveal that the Michael acceptor moiety is the key pharmacophore of the active hit 8a, and 8a is classified as the irreversible Michael acceptor accordingly.
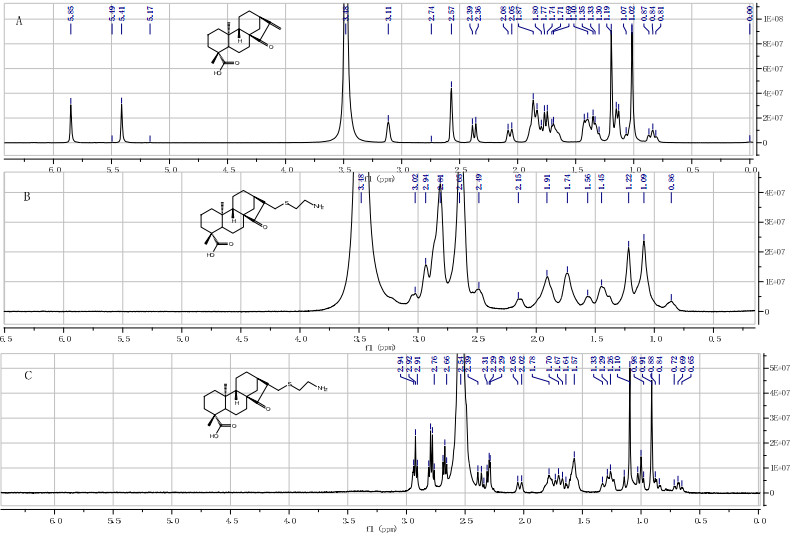
Reaction of compound 9a with cysteamine. A) 1H NMR spectrum of 9a in [D6]-DMSO; B) Spectrum recorded 5 min after the addition of 2 mol equiv cysteamine; C) Spectrum recorded 5 min after dilution (1:20) of the reaction mixture with CDCl3. All spectra were taken at 400 MHz. Note the complete and irreversible disappearance of the olefin signals at d= 5.49 and 5.41 ppm (H-17a and H-17b) upon addition of cysteamine.
In conclusion, a potential strategy to identify drug leads and re-discover the "trash" natural products is exemplified. The possible action mode of typical active hit 8a was also investigated. It was confirmed that the Michael acceptor moiety was the key pharmacophore and 8a was an irreversible thiol sink. And the superiority of the strategy is apparent, including resource saving, increased discovery rate, higher efficiency. Optimization process can be furthered based on the leads identified. Moreover, the strategy also provides an alternative view to evaluate the "trash" natural products, and may reward us serendipity.
Experimental Section
Material and Methods. Reagents and solvents were used as commercial grade. Toluene and dimethyl sulfoxide (DMSO) were treated as anhydrous solvents prior to use. Chromatographies were performed with 300–400 mesh silica gels. Thin layer chromatographies were carried out on Merck silica plates (0.25 mm layer thickness). ESIMS and HRESIMS were taken on a VG Auto Spec-3000 or on a Finnigan MAT 90 instrument. Optical rotations were measured with a Horiba SEPA-300 polarimeter. 1H and 13C NMR experiments were performed on a Bruker AM-300, AM-400 and DRX-500 NMR spectrometer at ambient temperature. And chemical shifts were given in δ with TMS as internal reference.
General Procedure for Preparation of Michael Acceptors 1a–14a16. To a solution of the substrate (1.0 mmol) in DMSO/toluene (0.8 mL/0.2 mL) was added IBX (3.0 mmol) in one portion. The mixture was heated to 85–95 ℃, and was followed by TLC until no starting material was detected. Then the mixture was cooled to room temperature and diluted with H2O. The aqueous layer was extracted with Et2O for three times. The combined organic layer was washed with 5% NaHCO3 (3 × 10 mL), H2O (1 × 10 mL), and dried (MgSO4), followed by removal of solvent in vacuo, leading to crude compounds which was purified using flash column chromatography on silica gel.
Oleana-1, 12-dien-3-oxo-28-oic acid (1a): white foam, 1H NMR (500 MHz, CDCl3): δ 7.06 (d, J = 10.1Hz, 1H), 5.81 (d, J = 10.1Hz, 1H), 5.31 (d, J = 3.7Hz, 1H), 2.23 (d, J = 11.2Hz, 1H), 2.17–2.13 (m, 1H), 2.09 (d, J = 3.9Hz, 1H), 2.05 (s, 1H), 2.01 (dd, J = 13.5, 4.2Hz, 1H), 1.87 (td, J = 13.5, 4.2Hz, 1H), 1.80 (dd, J = 11.4, 6.0Hz, 1H), 1.77–1.72 (m, 1H), 1.68 (dd, J = 13.2, 3.6Hz, 2H), 1.59–1.49 (m, 4H), 1.46–1.39 (m, 2H), 1.38–1.28 (m, 3H), 1.25 (d, J = 2.3Hz, 1H), 1.21 (dd, J = 6.4, 2.1Hz, 1H), 1.17 (s, 3H), 1.14 (s, 3H), 1.10 (s, 3H), 1.07 (s, 3H), 0.95 (d, J = 6.2Hz, 2H), 0.86 (s, 3H); 13CNMR (125MHz, CDCl3)δ 206.2 (C), 184.6 (C), 160.5 (CH), 139.9 (C), 126.4 (CH), 55.9 (C), 54.8 (CH), 45.9 (C), 43.7 (C), 43.0 (CH), 41.6 (C), 40.7 (C), 40.3 (CH), 40.1 (CH), 38.0 (CH2), 34.1 (CH2), 31.9 (CH2), 29.3 (CH2), 29.2 (CH3), 25.3 (CH2), 24.8 (CH3), 24.6 (CH2), 22.9 (CH3), 22.4 (CH2), 20.2 (CH2), 20.1 (CH3), 18.9 (CH3), 18.3 (CH3); HREIMS (m/z):452.3283[M]+ (calcdforC30H44O3, 452.3290).
Ursa-1, 12-dien-3-oxo-28-oic acid (2a): white foam, 1H NMR (400 MHz, CDCl3): δ 7.06 (d, J = 10.1Hz, 1H), 5.81 (d, J = 10.1Hz, 1H), 5.31 (d, J = 3.7Hz, 1H), 2.23 (d, J = 11.2Hz, 1H), 2.17–2.13 (m, 1H), 2.09 (d, J = 3.9Hz, 1H), 2.05 (s, 1H), 2.01 (dd, J = 13.5, 4.2Hz, 1H), 1.87 (td, J = 13.5, 4.2Hz, 1H), 1.80 (dd, J = 11.4, 6.0Hz, 1H), 1.77–1.72 (m, 1H), 1.68 (dd, J = 13.2, 3.6Hz, 2H), 1.59–1.49 (m, 4H), 1.46–1.39 (m, 2H), 1.38–1.28 (m, 3H), 1.25 (d, J = 2.3Hz, 1H), 1.21 (dd, J = 6.4, 2.1Hz, 1H), 1.17 (s, 3H), 1.14 (s, 3H), 1.10 (s, 3H), 1.07 (s, 3H), 0.95 (d, J = 6.2Hz, 2H), 0.86 (s, 3H); 13CNMR (100MHz, CDCl3)δ 205.3 (C), 183.5 (C), 159.2 (CH), 138.5 (C), 125.0 (CH), 53.3 (CH), 52.6 (CH), 48.0 (C), 44.4 (C), 43.0 (CH), 42.2 (C), 41.5 (CH), 40.1 (C), 39.3 (C), 38.8 (CH), 38.7 (CH), 36.5 (CH2), 32.6 (CH2), 30.6 (CH2), 27.8 (CH2), 27.8 (CH3), 23.9 (CH2), 23.4 (CH3), 23.1 (CH2), 21.5 (CH3), 21.0 (CH3), 18.7 (CH3), 17.5 (CH3), 16.9 (CH3); HREIMS (m/z):452.3291[M]+ (calcdforC30H44O3, 452.3290).
Lupa-1, 12, 20 (29)-trien-3-one (3a): white foam, 1H NMR (400 MHz, CDCl3): δ 7.09 (d, J = 10.2Hz, 1H), 5.78 (d, J = 10.2Hz, 1H), 4.70 (d, J = 1.7Hz, 1H), 4.58 (s, 1H), 2.39 (td, J = 11.0, 5.7Hz, 1H), 2.04 (s, 1H), 1.97–1.88 (m, 1H), 1.72 (d, J = 4.2Hz, 1H), 1.68 (s, 3H), 1.67–1.59 (m, 2H), 1.58 (s, 1H), 1.56–1.50 (m, 4H), 1.47 (dt, J = 9.4, 3.9Hz, 3H), 1.43–1.32 (m, 5H), 1.24 (d, J = 4.1Hz, 1H), 1.20 (d, J = 11.0Hz, 1H), 1.12 (s, 3H), 1.10 (s, 3H), 1.07 (s, 3H), 1.06 (s, 3H), 1.03–0.99 (m, 1H), 0.95 (s, 3H), 0.80 (s, 3H); 13CNMR (100MHz, CDCl3)δ 205.4 (C), 159.8 (CH), 150.6 (C), 125.1 (CH), 109.4 (CH2), 53.3 (CH), 48.0 (CH), 47.8 (CH), 44.5 (C), 44.3 (CH), 43.0 (C), 42.9 (C), 41.7 (C), 39.9 (CH2), 39.5 (C), 38.1 (CH), 35.4 (CH2), 33.7 (CH2), 29.7 (CH2), 27.7 (CH3), 27.3 (CH2), 25.0 (CH2), 21.3 (CH), 21.1 (CH2), 19.2 (CH), 19.1 (CH3), 18.9 (CH2), 17.9 (CH3), 16.4 (CH3), 14.3 (CH3); HREIMS (m/z):422.3548[M]+ (calcdforC30H46O, 422.3549).
Lupa-1, 12, 20 (29)-trien-3-oxo-28-oic acid (4a): white foam, 1H NMR (500 MHz, CDCl3): δ 7.11 (d, J = 10.3Hz, 1H), 5.80 (d, J = 10.2Hz, 1H), 4.76 (s, 1H), 4.64 (s, 1H), 3.03 (dd, J = 10.7, 6.0Hz, 1H), 2.34–2.28 (m, 1H), 2.26 (dd, J = 12.3, 3.2Hz, 1H), 2.03–1.96 (m, 2H), 1.79 (d, J = 13.2Hz, 1H), 1.70 (s, 3H), 1.65 (t, J = 11.4Hz, 2H), 1.60–1.56 (m, 1H), 1.56–1.50 (m, 4H), 1.44 (m, 5H), 1.26 (s, 6H), 1.13 (s, 3H), 1.07 (s, 3H), 1.06 (s, 3H), 1.02 (s, 3H), 1.00 (s, 3H), 0.88 (t, J = 6.7Hz, 1H); 13CNMR (125MHz, CDCl3)δ 205.4 (C), 181.5 (C), 160.5 (CH), 159.6 (CH), 150.1 (C), 125.1 (CH), 109.8 (CH2), 56.3 (C), 53.4 (CH), 49.1 (CH), 46.8 (CH), 44.6 (C), 44.5 (CH), 42.7 (C), 41.6 (C), 39.5 (C), 38.6 (CH), 37.0 (CH2), 33.8 (CH2), 32.1 (CH2), 30.5 (CH2), 29.6 (CH2), 29.6 (CH2), 27.7 (CH2), 25.4 (CH2), 21.3 (CH3), 21.1 (CH2), 19.3 (CH3), 19.1 (CH3), 18.9 (CH2), 16.4 (CH3), 14.5 (CH3); HREIMS (m/z):452.3282[M]+ (calcdforC30H44O3, 452.3290).
Dammar-1, 24 (25)-dien-3, 12-di-one (5a): white foam, 1H NMR (500 MHz, CDCl3): δ 6.94 (d, J = 10.3Hz, 1H), 5.77 (d, J = 10.2Hz, 1H), 5.04 (s, 1H), 2.85 (d, J = 10.3Hz, 1H), 2.44 (dd, J = 14.0, 4.0Hz, 1H), 2.35 (dt, J = 13.7, 6.6Hz, 2H), 1.95 (dd, J = 13.2, 3.9Hz, 2H), 1.77 (dd, J = 21.8, 11.4Hz, 1H), 1.62 (s, 3H), 1.57 (d, J = 2.6Hz, 1H), 1.55 (s, 3H), 1.41 (d, J = 13.4Hz, 2H), 1.32–1.23 (m, 2H), 1.22 (d, J = 4.1Hz, 1H), 1.20 (s, 3H), 1.16 (s, 1H), 1.10 (d, J = 3.1Hz, 6H), 1.05 (s, 6H), 1.00 (s, 1H), 0.97 (s, 1H); 13CNMR (125MHz, CDCl3)δ 212.0 (C), 204.5 (C), 157.1 (CH), 131.6 (C), 125.8 (CH), 124.7 (CH), 73.2 (C), 56.4 (CH), 54.8 (C), 53.8 (CH), 47.4 (CH), 45.7 (CH), 44.7 (C), 41.0 (C), 39.7 (C), 39.0 (CH2), 38.2 (CH2), 33.4 (CH2), 30.8 (CH2), 27.6 (CH3), 26.4 (CH3), 25.7 (CH3), 24.5 (CH2), 22.5 (CH2), 21.3 (CH3), 19.0 (CH2), 18.9 (CH3), 17.6 (CH3), 17.3 (CH3), 16.1 (CH3); HREIMS (m/z):454.3453[M]+ (calcdforC30H46O3, 454.3447).
9, 19-Cyclolanost-1, 24 (25)-dien-3-oxo-26-oic acid (6a): white foam, 1H NMR (500 MHz, CDCl3): δ 6.90 (t, J = 7.4Hz, 1H), 6.79 (d, J = 10.1Hz, 1H), 6.73 (s, OH), 5.96 (d, J = 10.0Hz, 1H), 2.12 (dd, J = 9.9, 6.0Hz, 2H), 2.02–1.95 (m, 1H), 1.90 (dd, J = 12.9, 7.4Hz, 2H), 1.84 (s, 3H), 1.71–1.49 (m, 7H), 1.44 (d, J = 9.2Hz, 1H), 1.36–1.23 (m, 6H), 1.10 (s, 3H), 0.96 (s, 6H), 0.92 (d, J = 6.4Hz, 3H), 0.89 (s, 3H), 0.76 (d, J = 4.6Hz, 1H); 13CNMR (125MHz, CDCl3)δ 205.4 (C), 173.3 (C), 154.1 (CH), 145.6 (CH), 126.6 (CH), 126.6 (C), 51.8 (CH), 49.1 (C), 45.9 (C), 45.3 (C), 44.2 (CH), 43.5 (CH), 35.9 (CH), 34.7 (CH2), 34.4 (CH2), 32.2 (CH2), 29.8 (C), 29.1 (CH2), 27.8 (CH2), 27.7 (CH2), 25.8 (CH2), 24.7 (C), 23.2 (CH2), 21.3 (CH3), 19.4 (CH2), 19.0 (CH3), 18.4 (CH3), 18.1 (CH3), 16.8 (CH3), 11.9 (CH3); HREIMS (m/z):452.3289[M]+ (calcdforC30H44O3, 452.3290).
18-Norpregnane-1-en-3, 12, 20-tri-one (7a): white foam, 1H NMR (500 MHz, CDCl3): δ 6.99 (d, J = 10.2Hz, 1H), 5.82 (d, J = 10.2Hz, 1H), 3.22 (td, J = 10.7, 6.9Hz, 1H), 3.12 (d, J = 10.5Hz, 1H), 2.44 (d, J = 4.4Hz, 1H), 2.41 (d, J = 13.1Hz, 1H), 2.22 (s, 3H), 2.21 (s, 1H), 2.12–2.05 (m, 1H), 1.97 (dd, J = 12.8, 4.4Hz, 1H), 1.83 (dd, J = 20.0, 10.9Hz, 1H), 1.73–1.67 (m, 2H), 1.65 (d, J = 2.8Hz, 2H), 1.61 (d, J = 2.2Hz, 1H), 1.47 (d, J = 12.7Hz, 1H), 1.30 (d, J = 9.8Hz, 1H), 1.26 (s, 3H), 1.23 (s, 2H), 1.15 (s, 3H), 1.14 (s, 3H), 1.10 (s, 3H); 13CNMR (125MHz, CDCl3)δ 210.4 (C), 208.5 (C), 204.6 (C), 157.3 (CH), 125.7 (CH), 58.0 (CH), 54.3 (C), 53.7 (CH), 47.2 (CH), 46.9 (CH), 44.7 (C), 40.9 (C), 39.7 (C), 38.5 (CH2), 33.6 (CH2), 31.2 (CH2), 30.1 (CH3), 29.6 (CH2), 27.6 (CH3), 25.6 (CH2), 21.3 (CH3), 18.9 (CH2), 18.9 (CH3), 17.0 (CH3), 16.0 (CH3); HREIMS (m/z):370.2516[M]+ (calcdforC24H34O3, 370.2516).
Kaur-16-en-15-oxo-18-oic acid (8a): white foam, 1H NMR (500 MHz, CDCl3): δ 5.78 (s, 1H), 5.14 (s, 1H), 2.91 (s, 1H), 2.28 (d, J = 11.9Hz, 1H), 2.00 (d, J = 13.1Hz, 1H), 1.77 (dd, J = 11.5, 7.9Hz, 1H), 1.70 (ddd, J = 23.8, 12.4, 6.8Hz, 5H), 1.54 (dd, J = 15.4, 5.8Hz, 2H), 1.29 (ddd, J = 16.6, 13.1, 6.0Hz, 3H), 1.21 (dd, J = 10.3, 3.2Hz, 1H), 1.08 (s, 3H), 1.03–0.98 (m, 1H), 0.88 (s, 3H); 13CNMR (125MHz, CDCl3)δ 211.6 (C), 180.3 (C), 149.3 (C), 114.7 (CH), 55.8 (CH), 52.4 (C), 51.5 (CH), 43.2 (C), 40.0 (C), 39.8 (CH2), 37.8 (CH), 37.6 (CH2), 36.2 (CH2), 33.4 (CH2), 31.8 (CH2), 28.6 (CH3), 19.8 (CH2), 18.6 (CH2), 18.1 (CH2), 15.2 (CH3); HREIMS (m/z):316.2039[M]+ (calcdforC20H28O3, 316.2039).
Kaur-16-en-11, 15-dioxo-18-oic acid (9a): 1H NMR (500MHz, MeOD)δ 6.03 (s, 1H), 5.42 (s, 1H), 3.28 (s, 1H), 2.82 (d, J = 12.5Hz, 1H), 2.65 (dd, J = 12.1, 4.0Hz, 1H), 2.57 (d, J = 15.9Hz, 1H), 2.20 (d, J = 9.6Hz, 1H), 2.02 (m, 1H), 1.97 (d, J = 14.0Hz, 1H), 1.92 (s, 1H), 1.89 (d, J = 3.6Hz, 1H), 1.88 (s, 1H), 1.80 (d, J = 13.9Hz, 1H), 1.70–1.76 (m, 2H), 1.59 (d, J = 10.2Hz, 1H), 1.48 (d, J = 14.7Hz, 1H), 1.29 (s, 3H), 1.23 (s, 1H), 1.20 (dd, J = 13.6, 4.3Hz, 1H), 1.09 (dd, J = 13.5, 4.1Hz, 1H), 1.02 (s, 3H), 0.88 (m, 1H); 13CNMR (125MHz, CDCl3)δ 209.1 (C), 206.5 (C), 183.4 (C), 147.4 (C), 118.5 (CH2), 67.0 (CH), 55.4 (CH), 52.64 (C), 51.4 (CH2), 43.8 (C), 40.0 (C), 37.4 (CH2), 36.5 (CH), 35.8 (CH2), 32.0 (CH2), 29.6 (CH2), 28.8 (CH3), 19.8 (CH2), 18.5 (CH2), 17.1 (CH3); HREIMS (m/z):330.1833[M]+ (calcdforC20H26O4, 330.1831).
Labda-1, 11-dien-3-oxo-13-oic acid (10a): white foam, 1H NMR (500 MHz, CDCl3): δ 7.10 (d, J = 10.4Hz, 1H), 5.97 (d, J = 10.4Hz, 1H), 5.71 (s, 1H), 5.03 (s, 1H), 4.69 (s, 1H), 2.51 (d, J = 13.0Hz, 1H), 2.21 (s, 3H), 2.17 (s, 1H), 1.90 (dd, J = 20.1, 6.9Hz, 2H), 1.73 (ddd, J = 17.8, 15.0, 8.4Hz, 2H), 1.53 (dd, J = 13.0, 3.9Hz, 1H), 1.16 (s, 3H), 1.06 (s, 3H), 0.92 (s, 3H).; 13CNMR (125MHz, CDCl3)δ 204.6 (C), 171.4 (C), 162.6 (C), 155.7 (CH), 146.0 (C), 126.9 (CH), 115.1 (CH), 109.3 (CH2), 52.3 (CH), 51.3 (CH), 44.7 (C), 29.6 (C), 27.0 (CH3), 24.0 (CH2), 22.0 (CH2), 21.8 (CH3), 19.2 (CH3), 16.3 (CH3); HREIMS (m/z):288.1724[M]+ (calcdforC18H24O3, 288.1725).
8, 14-Secogammacera-1, 8, 14, 19-tetra-en-3, 21-di-one (11a): white foam, 1HNMR (500MHz, CDCl3): δ 7.10 (d, J = 10.4Hz, 1H), 5.97 (d, J = 10.4Hz, 1H), 5.71 (s, 1H), 5.03 (s, 1H), 4.69 (s, 1H), 2.51 (d, J = 13.0Hz, 1H), 2.21 (s, 3H), 2.17 (s, 1H), 1.90 (dd, J = 20.1, 6.9Hz, 2H), 1.73 (ddd, J = 17.8, 15.0, 8.4Hz, 2H), 1.53 (dd, J = 13.0, 3.9Hz, 1H), 1.16 (s, 3H), 1.06 (s, 3H), 0.92 (s, 3H).; 13CNMR (125MHz, CDCl3)δ 204.6 (C), 171.4 (C), 162.6 (C), 155.7 (CH), 146.0 (C), 126.9 (CH), 115.1 (CH), 109.3 (CH2), 52.3 (CH), 51.3 (CH), 44.7 (C), 29.6 (C), 27.0 (CH3), 24.0 (CH2), 22.0 (CH2), 21.8 (CH3), 19.2 (CH3), 16.3 (CH3); HREIMS (m/z):342.1468[M]+ (calcdforC20H22O5, 342.1467).
8, 14-Secogammacera-1, 8, 14, 19-tetra-en-3, 21-di-one (12a): white foam, 1H NMR (500 MHz, CDCl3): δ 7.04 (d, J = 10.3Hz, 1H), 5.96 (d, J = 10.3Hz, 1H), 5.06 (s, 1H), 4.79 (s, 1H), 2.55 (d, J = 12.9Hz, 1H), 2.09 (t, J = 10.6Hz, 1H), 1.97–1.84 (m, 2H), 1.80 (d, J = 10.1Hz, 2H), 1.61 (s, 2H), 1.55 (dd, J = 13.0, 4.1Hz, 1H), 1.48 (d, J = 7.4Hz, 1H), 1.27 (s, 2H), 1.18 (s, 3H), 1.07 (s, 3H), 0.90 (s, 3H).; 13CNMR (125MHz, CDCl3)δ 204.7 (C), 156.1 (CH), 146.5 (C), 126.8 (CH), 109.4 (CH2), 52.3 (CH), 52.2 (CH), 44.6 (C), 41.4 (C), 37.7 (CH2), 29.6 (C), 27.0 (CH3), 24.0 (CH2), 22.8 (CH2), 21.7 (CH3), 16.4 (CH3); HREIMS (m/z):434.3173[M]+ (calcdforC30H42O2, 434.3173).
Stigmasta-1, 5-dien-3-one (13a): white foam, 1H NMR (400 MHz, CDCl3): δ 6.15 (d, J = 11.0Hz, 1H), 6.09 (dd, J = 9.8, 2.4Hz, 1H), 5.67 (s, 1H), 2.60–2.55 (m, 1H), 2.54 (d, J = 5.3Hz, 1H), 2.45 (d, J = 3.7Hz, 1H), 2.41 (d, J = 5.6Hz, 1H), 2.19 (t, J = 10.3Hz, 1H), 2.08 (d, J = 3.2Hz, 1H), 2.06–2.03 (m, 1H), 2.03–1.97 (m, 1H), 1.95–1.87 (m, 1H), 1.73–1.69 (m, 1H), 1.69–1.65 (m, 1H), 1.61 (s, 3H), 1.55–1.50 (m, 1H), 1.45 (d, J = 3.7Hz, 1H), 1.40 (dd, J = 13.0, 3.7Hz, 2H), 1.34 (d, J = 12.8Hz, 2H), 1.30 (d, J = 7.7Hz, 2H), 1.27–1.13 (m, 12H), 1.11 (s, 3H), 1.05–0.99 (m, 2H), 0.94 (t, J = 6.3Hz, 3H), 0.87 (s, 3H), 0.85 (s, 3H), 0.83 (d, J = 2.3Hz, 3H), 0.81 (d, J = 2.7Hz, 3H), 0.76 (d, J = 4.6Hz, 3H), 0.71 (s, 1H); 13CNMR (100MHz, CDCl3)δ 199.7 (C), 164.0 (C), 141.6 (CH), 127.7 (CH), 123.4 (CH), 55.8 (CH), 53.3 (CH), 50.6 (CH), 45.7 (CH), 43.3 (C), 39.4 (CH2), 37.7 (CH), 36.1 (CH), 33.9 (CH2), 33.8 (CH2), 33.8 (CH2), 29.0 (CH), 28.1 (CH2), 26.0 (CH2), 23.6 (CH2), 23.0 (CH2), 20.6 (CH2), 19.7 (CH3), 18.9 (CH3), 18.6 (CH3), 16.2 (CH3), 11.9 (CH3), 11.8 (CH3); HREIMS (m/z):410.3546[M]+ (calcdforC29H46O, 410.3549).
Stigmasta-4-methyl-1, 7 (8), 24 (28)-triene-3, 6, 23-tri-one (14a):white foam, 1H NMR (400 MHz, CDCl3):δ 6.15 (d, J = 11.0Hz, 1H), 6.09 (dd, J = 9.8, 2.4Hz, 1H), 5.67 (s, 1H), 2.60–2.55 (m, 1H), 2.54 (d, J = 5.3Hz, 1H), 2.45 (d, J = 3.7Hz, 1H), 2.41 (d, J = 5.6Hz, 1H), 2.19 (t, J = 10.3Hz, 1H), 2.08 (d, J = 3.2Hz, 1H), 2.06–2.03 (m, 1H), 2.03–1.97 (m, 1H), 1.95–1.87 (m, 1H), 1.73–1.69 (m, 1H), 1.69–1.65 (m, 1H), 1.61 (s, 3H), 1.55–1.50 (m, 1H), 1.45 (d, J = 3.7Hz, 1H), 1.40 (dd, J = 13.0, 3.7Hz, 2H), 1.34 (d, J = 12.8Hz, 2H), 1.30 (d, J = 7.7Hz, 2H), 1.27–1.13 (m, 12H), 1.11 (s, 3H), 1.05–0.99 (m, 2H), 0.94 (t, J = 6.3Hz, 3H), 0.87 (s, 3H), 0.85 (s, 3H), 0.83 (d, J = 2.3Hz, 3H), 0.81 (d, J = 2.7Hz, 3H), 0.76 (d, J = 4.6Hz, 3H), 0.71 (s, 1H); 13CNMR (100MHz, CDCl3)δ 202.7 (C), 200.7 (C), 198.1 (C), 160.4 (C), 152.2 (CH), 148.5 (C), 135.1 (CH), 127.3 (CH), 123.8 (CH), 55.8 (CH), 53.3 (CH), 50.6 (CH), 45.7 (CH), 43.3 (C), 39.4 (CH2), 37.7 (CH), 36.1 (CH), 33.9 (CH2), 33.8 (CH2), 33.8 (CH2), 29.0 (CH), 28.1 (CH2), 26.0 (CH2), 23.6 (CH2), 23.0 (CH2), 20.6 (CH2), 19.7 (CH3), 18.9 (CH3), 18.6 (CH3), 16.2 (CH3), 11.9 (CH3), 11.8 (CH3); HREIMS (m/z):450.3136[M]+ (calcdforC30H42O3, 450.3134).
Thiol Sensitive Assay. Compound 8a (10 mg, 0.03 mmol) was dissolved in d6-DMSO (500 μL) in a 5 mm NMR tube, and the spectrum was recorded (Figure 2A). Cysteamine (4.7 mg, 0.06 mmol, 2 mol equiv) was then added, and the spectrum was recorded 5 min after the addition (Figure 2B). An aliquot (25 μL) of the solution was then transferred into a second NMR tube containing CDCl3 (500 μL) and a new spectrum was recorded (Figure 2C). A positive result was evidenced by the disappearance of a particular olefin system of the substrate, and the irreversibility of the Michael addition by its disappearance upon dilution 1:20 with CDCl3.
Biological Assay. The used cell lines were human promyelocytic leukemia cell line (HL-60), human hepatocellular carcinoma cell line (SK-BR-3), human lung carcinoma cell line (A-549), human breast adenocarcinoma cell line (SMMC-7721), human pancreatic carcinoma cell line (PANC-1). An MTT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide] colorimetric assay was performed in 96-well plates. HL-60 cells at the log phase of their growth cycle (1.25 × 105 cell/mL) were added to each well (90 mL/well), then treated in four replicates at various concentrations of the samples (1–100 µg/mL), and incubated for 48 hours at 37 ℃ in a humidified atmosphere of 5% CO2. After 48 h, 10 µL of MTT solution (5 mg/mL) per well was added to each cultured medium, which were incubated for further 4 hours. Then, a three-system solution of 10% SDS–5% isobutanole–0.012 mol/L hydrochloric acid was added to each well (100 µL/well). After 12 h at room temperature, the OD of each well was measured on a Microplate Reader (BIO-TEK instruments Inc EL311S) at a wavelength of 570 nm. In these experiments, the negative reference agents was 0.1% DMSO, and cisplatin (DDP) was used as the positive control with concentration of 1–80 µg/mL. The same method was used in cytotoxic testing against SKBR-3, A549, SMMC-7721 and PANC-1 cell lines.
Notes
Electronic Supplementary Material
Supplementary material is available in the online version of this article at http://dx.doi.org/ 10.1007/s13659-012-0071-7 and is accessible for authorized users.
Acknowledgments
We thanked Mrs. Li-Yan Peng, Dr. Juan He, Dr. ZhengHong Pan, Dr. Xing-De Wu and Mr. Liao-Bin Dong for generously providing samples of natural products. We thanked the National Natural Science Foundation of China (No. 90813004, U0932602, 20802083 and 973 Program No. 2009CB522303 and No. 2011CB915503) and the State Key Laboratory of Phytochemistry and Plant Resources in West China (P2010-ZZ18) for financial support.
References
-
1.D. Fattori, A. Squarcia, S. Bartoli, Drugs in R&D 9, 217-227 (2008) PubMed Google Scholar
-
2.M. Xiang, Y. Cao, W. Fan, L. Chen, Y. Mo, Comb. Chem. High Throughput Screening 15, 328-337 (2012) CrossRef PubMed Google Scholar
-
3.J. Mestres, G. H. Veeneman, J. Med. Chem. 46, 3441 (2003) CrossRef PubMed Google Scholar
-
4.P. Gribbon, S. Andreas, Drug Discov. Today 10, 17-22 (2005) CrossRef PubMed Google Scholar
-
5.J. Clardy, C. Walsh, Nature 432, 829-837 (2004) CrossRef PubMed Google Scholar
-
6.D. J. Newman, G. M. Cragg, J. Nat. Prod. 70, 461-477 (2007) PubMed Google Scholar
-
7.S. Danishefsky, Nat. Prod. Rep. 27, 1114-1116 (2010) CrossRef PubMed Google Scholar
-
8.A. O. Aptula, D. W. Roberts, Chem. Res. Toxicol. 19, 1097-1105 (2006) CrossRef PubMed Google Scholar
-
9.S. Amslinger, ChemMedChem 5, 351-356 (2010) CrossRef PubMed Google Scholar
-
10.L. Garuti, M. Roberti, G. Bottegoni, Curr. Med. Chem. 18, 2981-2994 (2011) CrossRef PubMed Google Scholar
-
11.R. D. Couch, R. G. Browning, T. Honda, G. W. Gribble, D. L. Wright, M. B. Sporn, A. C. Anderson, Bioorg. Med. Chem. Lett. 15, 2215-2219 (2005) CrossRef PubMed Google Scholar
-
12.A. J. Barrett, N. D. Rawlings, E. A. O'Brien, J. Struct. Biol. 134, 95-102 (2001) CrossRef PubMed Google Scholar
-
13.L. Leloup, A. Wells, Expert Opin. Ther. Targets 15, 309-323 (2011) CrossRef PubMed Google Scholar
-
14.H. H. Otto, T. Schirmeister, Chem. Rev. 97, 133-172 (1997) CrossRef PubMed Google Scholar
-
15.A. Petronelli, G. Pannitteri, U. Testa, Anti-Cancer Drugs 20, 880-892 (2009) CrossRef PubMed Google Scholar
-
16.A. T. Dinkova-Kostova, K. T. Liby, K. K. Stephenson, W. D. Holtzclaw, X. Gao, N. Suh, C. Williams, R. Risingsong, T. Honda, G. W. Gribble, M. B. Sporn, P. Talalay, Proc. Natl. Acad. Sci. USA 102, 4584-4589 (2005) CrossRef PubMed Google Scholar
-
17.B. R. Shenai, B. J. Lee, A. Alvarez-Hernandez, P. Y. Chong, C. D. Emal, R. J. Neitz, W. R. Roush, P.J. Rosenthal, Antimicrob. Agents Ch. 47, 154-160 (2003) CrossRef PubMed Google Scholar
-
18.O. D. Ekici, Z. Z. Li, A. J. Campbell, K. E. James, J. L. Asgian, J. Mikolajczyk, G. S. Salvesen, R. Ganesan, S. Jelakovic, M. G. Gruetter, J. C. Powers, J. Med. Chem. 49, 5728-5749 (2006) CrossRef PubMed Google Scholar
-
19.J. Schust, B. Sperl, A. Hollis, T. U. Mayer, T. Berg, Chem. Biol. 13, 1235-1242 (2006) CrossRef PubMed Google Scholar
-
20.Y. Ito, T. Hirao, T. Saegusa, J. Org. Chem. 43, 1011-1013 (1978) CrossRef PubMed Google Scholar
-
21.H. J. Reich, J. M. Renga, I. L. Reich, J. Am. Chem. Soc. 97, 5434-5447 (1975) CrossRef PubMed Google Scholar
-
22.K. C. Nicolaou, T. Montagnon, P. S. Baran, Y. L. Zhong, J. Am. Chem. Soc. 124, 2245-2258 (2002) CrossRef PubMed Google Scholar
-
23.C. Avonto, O. Taglialatela-Scafati, F. Pollastro, A. Minassi, V. Di Marzo, L. De Petrocellis, G. Appendino, Angew. Chem. Int. Ed. 50, 467-471 (2011) CrossRef PubMed Google Scholar
-
24.J. L. Torres, C. Lozano, L. Julià, F. J. Sánchez-Baeza, J. M. Anglada, J. J. Centelles, M. Cascante, Bioorg. Med. Chem. 10, 2497-2509 (2002) CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2012
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
