


Rauvotetraphyllines A–E, new indole alkaloids from Rauvolfia tetraphylla
Abstract
Five new indole alkaloids rauvotetraphyllines A-E(1-5), together with eight known analogues, were isolated from the aerial parts of Rauvolfia tetraphylla. The structures were established by means of spectroscopic methods.Keywords
Rauvolfia tetraphylla indole alkaloid rauvotetraphyllineIntroduction
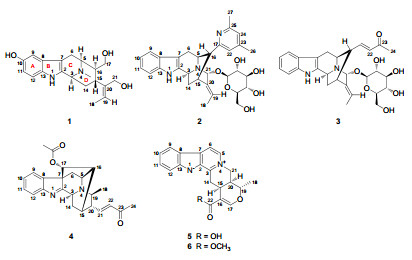
Rauvolfia genus, family Apocynaceae, continues to be fascinating as it produces novel heterocyclic alkaloids with monoterpene indole skeletons, which have attracted great interests from biological and therapeutic aspects, due to their anticancer, 1 antimalarial, 2 antihypertensive, 3 and sedative4 properties. The isolation and structure elucidation of three indole alkaloids rauvoyunines A, B, and C from the aerial parts of Rauvolfia yunnanensis have been reported earlier.5 As one part of our ongoing research program exploring bioactive indole alkaloids from Chinese species of Rauvolfia, phytochemical analysis has been carried out on the aerial parts of R. tetraphylla collected from Yunnan Province, with particular attention to the monoterpene indole constituents, and has resulted in the isolation of five new alkaloids, rauvotetraphyllines A–E (1–5), as well as eight known analogues, alstonine (6), 6 nortetraphyllicine, 7 peraksine, 8 sarpagine, 9 3-hydroxysarpagine, 10 dihydroperaksine, 11 10-hydroxydihydroperaksine, 11 and raucaffricine.12 The present paper reports the isolation, structure elucidation, and cytotoxic evaluation of the new compounds.
|
Results and Discussion
Compound 1, obtained as amorphous powder, had a molecular formula of C20H26N2O3 based on HRESIMS (pos.) at m/z 343.2024 (calcd for C20H27N2O3, 343.2021). In the UV spectrum, two noticeable maxima absorption bands at 211 and 275 nm as well as two shoulders at 222 and 310 nm were detected, suggesting the existence of an O-substituted indole chromophore.13 The IR spectrum showed band at 3407 cm–1, due to OH/NH functionalities. The 1H NMR spectrum (Table 1) showed signals for an aromatic AMX spin system at δH 6.62 (dd, J = 8.5, 2.0 Hz), 6.82 (d, J = 2.0 Hz), and 7.11 (d, J = 8.5 Hz), typical of an indole moiety substituted by a hydroxy group at C-10 or C-11 position, signals characteristic of an ethylidene group at δH 1.16 (d, J = 6.3 Hz) and 5.51 (q, J = 6.3 Hz), protons for N-methyl group at δH 2.47 (s), and protons of two oxygenated methylene groups, one at δH 3.57 (dd, J = 11.1, 4.0 Hz) and 3.32 (dd, J = 11.1, 10.6 Hz), and another at δH 4.04 and 4.08 (each d, J = 13.4 Hz). In addition to resonances due to indole chromophore including an oxygen-bearing carbon at δC 151.2 (s), the 13C NMR (DEPT) spectrum (Table 2) exhibited signals ascribed to one double bond at δC 123.6 (d) and 140.6 (s), two oxygenated carbons at δC 62.9 (t) and δC 63.2 (t), two methyl signals at δC 12.7 (q) and 41.7 (q), and the other six aliphatic carbons. These spectroscopic features resembled rauvoyunine A, 5 a macroline-type alkaloid recently isolated from R. yunnanensis, except for the absence of an N-methyl group in ring B, as well as discrepant coupling constants in ring D. The position of the N-methyl group was confirmed by HMBC correlations from N(4)-Me to C-3 and C-5, while the location of the hydroxy group was confirmed by HMBC correlation from an m-coupling doublet of H-9 at δH 6.82 (d, J = 2.0 Hz) to C-7 and ROESY correlation between H-9 and H-6β (Figure 1). Since the J5, 6, J3, 14, and J14, 15 values corresponding to vicinal interaction (3JH, H) were essentially unchanged compared to those of rauvoyunine A, it is safe to deduce that the C/D ring junction stereochemistry remained intact, whereas revealing as the major difference from rauvoyunine A, the J values between H-15 and H-16 (1: J = 11.8 Hz; rauvoyunine A: J = 5.6 Hz), H-5 and H-16 (1: J = 5.0 Hz; rauvoyunine A: J ≈ 0 Hz), and H-16 and H2-17 (1: J = 10.6 and 4.0 Hz; rauvoyunine A: J = 5.5 and 3.7 Hz) strongly suggested that this new compound had a macroline-type skeleton with the unique feature of a 16-epi (16αH) form. Consistent with this deduction, the ROESY spectrum of 1 showed cross-peaks of H-15↔H2-17 and H2-17↔H-6β. The C-19–C-20 double bond had the E-configuration since the ROESY correlations of Me-18↔H-15 and H-19↔H2-21 were observed. Thus, the structure of the new alkaloid was established as shown, named rauvotetraphylline A.
1H NMR data for compounds 1−5 (δ in ppm, J in Hz)
13C NMR data for compounds 1−5 (δ in ppm)
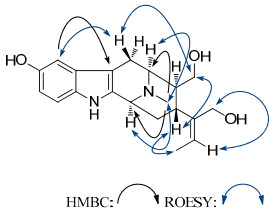
Key HMBC and ROESY correlations of 1
Compound 2, obtained as amorphous powder, possessed a molecular formula of C31H37N3O6, as evidenced by HRESIMS (pos.) at m/z 548.2768, in combination with NMR spectra (Tables 1 and 2), requiring 15 degrees of unsaturation. The NMR spectra exhibited a β-glucopyranosyl unit (δC 102.8, 75.3, 78.1, 71.5, 78.0, 62.8), and a set of resonances [δC 121.0 (d), 150.1 (s), 123.6 (d), 158.4 (s), 21.0 (q), 23.6 (q); δH 7.02 (s), 6.96 (s), 2.30 (s), 2.39 (s)] which can be easily determined as a 4, 6-dimethyl-2-pyridyl moiety.14 The remainder were closely related to those of vellosimine15—a sarpagine-type alkaloid, except for the absence of an aldehyde group, as well as an unusual low-field methine signal at δC 91.8 instead of a methylene signal for C-21. The significant downfield shift for C-21 suggested that the sugar unit was adjacent to C-21, as supported by HMBC correlation from the anomeric proton of β-glucose at δH 4.70 (d, J = 7.8 Hz) to C-21. Considering that it lacks an aldehyde group at C-16, which is usual for the sarpagine series such as 12-methoxy-vellosimine16 or dihydroperaksine-17-al11 from species of Rauvolfia, the 4, 6-dimethyl-2-pyridyl moiety was assumed to be an artifact (produced by aldehyde-ammonia condensation reaction) linked to C-16 since acetone and NH3·H2O were used as eluent during the isolation process. The above deduction was confirmed by HMBC correlations from H-5 and H-15 to C-17, and H-16 to C-17 and C-22. ROESY correlations of H-5↔H-6α(strong), H-5↔H6β(slight), H-3↔H-5 (slight), H-6β↔H-16, and H-16↔H-14βsuggested that the new alkaloid had the same ring junction stereochemistry as that of vellosimine, while the proton at C-22 showing cross-peaks with H-5, H-19, and H-21 supported exo position of the pyridyl moiety and β-orientation of H-21. The Me-18 at δH 1.15 (noticeable upfield shift being due to shielding by the pyridyl ring current) showed ROESY correlation with H-15 while H-19 exhibited correlation with H-21, revealing an E-geometry of the ethylidene. Therefore, the structure of 2 was unambiguously elucidated as shown, named rauvotetraphylline B.
Compound 3 was obtained as amorphous powder. High resolution mass spectrometry revealed the [M + H]+ peak at m/z 511.2431, suggesting the molecular formula C28H34N2O7. The NMR signals (Tables 1 and 2) were very similar to those of rauvotetraphylline B (2), which revealed that 3 was also a sarpagine alkaloid. However, there was a prominent difference as follows: the signals assigned to 4, 6-dimethyl-2-pyridyl moiety in 2 were absent, and there was a set of newly risen resonances: δC 151.7 (d), 131.6 (d), 201.1 (s), 27.0 (q); δH 6.73 (dd, J = 15.8, 9.1 Hz), 6.06 (d, J = 15.8 Hz), and 2.22 (s), which was easily assigned as an E-3-oxo-1-butenyl unit. In the HMBC spectrum, significant correlations from olefinic proton at δH 6.73 to carbons at δC 54.9 (d, C-5), 32.5 (d, C-15), and 46.8 (d, C-16) were observed, which indicated that the butenyl group was also attached to C-16. Other structural parts of 3 were identical to those of 2, as indicated by the HMBC, HSQC, and ROESY spectra. Consequently, the structure of 3 was determined as shown, named rauvotetraphylline C.
Compound 4 was isolated as amorphous powder. Its molecular formula was determined as C24H26N2O3 by the positive HRESIMS ([M + H]+ at m/z 391.2019). The NMR data (Tables 1 and 2) were analogous to those of perakine17—a ajmaline-type alkaloid. The principal difference between them was the aldehyde group in perakine changing into an E-3-oxo-1-butenyl unit [δC 146.0 (d), 131.7 (d), 198.0 (s), 27.4 (q); δH 6.84 (dd, J = 15.9, 7.8 Hz), 6.18 (d, J = 15.9 Hz), 2.28 (s)] on the basis of HMBC correlations from H-21 at δH 6.84 to C-15, C-19, and C-20. The ROESY correlations of H-3↔H-19↔H-14α, H-14β↔H-17, and Me-18↔H-20 indicated that 4 possessed the same stereochemical characteristics as that of perakine. Hence, the structure of 4 was assigned as shown, named rauvotetraphylline D.
Compound 5 and alstonine (6) exhibited nearly the same UV spectrum, with strong absorption maxima at 252, 308, and 370 nm. TLC analysis displayed spots with a clear blue fluorescence at 365 nm, between which 5 exhibited higher polarity. The molecular formula of 5 was determined as C20H18N2O3 by HRESIMS, 14 mass units lower than that of 6. The 1D NMR data of 5 (Tables 1 and 2) have similar chemical shifts and the same multiplicity as given for 6, except for a carboxyl group (δC 170.3) in 5 instead of the methoxycarbonyl group in 6, as supported by the lack of the methyl ester signals in the 1H and 13C NMR spectra and the presence of a hydroxy group as IR absorption band at 3423 cm–1 revealed. Other structural parts of 5 were identical to those of 6, as indicated by the HMBC, HSQC, and ROESY spectra. Thus, the structure of the alstonine derivative was established as shown, named rauvotetraphylline E.
All of the new alkaloids were evaluated for their cytotoxicity in vitro against five human cancer cell lines (HL-60, SMMC-7721, A-549, MCF-7, and SW-480) using the MTT method as reported previously.18 However, all tested compounds were inactive, and they showed IC50 values > 40μM. Considering the structural characteristics of the new compounds, we thought that 2–4 might have been formed as artifacts during isolation process.
Experimental Section
General Experimental Procedures. Optical rotations were measured on a Jasco P-1020 automatic digital polarimeter. IR spectra were obtained using a Bruker Tensor 27 FT-IR spectrometer with KBr pellets. UV data were obtained from online HPLC analysis. NMR spectra were acquired with a Bruker DRX-500 instrument at room temperature. ESIMS (including HRESIMS) were measured on API QSTAR Pulsar i mass spectrometers. Silica gel (200−300 mesh, Qingdao Marine Chemical Inc., China), Sephadex LH-20 (Amersham Biosciences, Sweden) and MCI gel CHP 20P (75−150 μm, Mitsubishi Chemical Corp., Tokyo, Japan) were used for column chromatography. Medium pressure liquid chromatography (MPLC) was performed on a Büchi Sepacore System equipping pump manager C-615, pump modules C-605, and fraction collector C-660 (Büchi Labortechnik AG, Switzerland), and columns packed with Chromatorex C-18 (40−75 μm, Fuji Silysia Chemical Ltd., Japan). Fractions were monitored by TLC (Qingdao Marine Chemical Inc., China) in combination with reversed-phase HPLC (Agilent 1200, Extend-C18 column, 5 μm, 4.6 × 150 mm).
Plant Material. The aerial parts of R. tetraphylla were collected in Xiaomenglun of Yunnan Province, China, in June 2010 and were identified by Prof. Yu Chen of Kunming Institute of Botany, Chinese Academy of Sciences. The voucher specimen was deposited at BioBioPha Co., Ltd.
Extraction and Isolation. The air-dried and powdered aerial parts of R. tetraphylla (7.5 kg) were extracted three times with 95% ethanol (3 × 50 L, each 3 days) at room temperature and filtered. The filtrate was evaporated under reduced pressure to get a residue (ca. 400 g), which was fractionized by silica gel column chromatography, eluted with a gradient solvent system of petroleum ether-acetone and then MeOH to yield six fractions A–F. Fraction F, eluted by MeOH, was separated on silica gel CC (CHCl3-MeOH, 50:1 → 0:1, v/v) to give five subfractions (F1–F5), Compounds 4 (6 mg) and 7 (7 mg) were obtained from the subfraction F1 by silica gel (CHCl3-MeOH-NH3·H2O, 50:1:0.1) and Sephadex LH-20 (MeOH) columns. Fraction F2 was separated by silica gel (CHCl3-MeOH-NH3·H2O, 40:1:0.1 → 5:1:0.1), MCI (50% → 100% MeOH in water), and Sephadex LH-20 (MeOH) columns to afford 8 (230 mg), 9 (94 mg), and 10 (84 mg). In the same way, 1 (119 mg) and 11 (29 mg) were isolated from fraction F3. Also, 12 (83 mg) and 6 (3 mg) were obtained from fractions F4. After repeated silica gel (CHCl3-MeOHNH3·H2O, 10:1:0.1 → 1:1:0.05), Sephadex LH-20 (CHCl3-MeOH, 1:1), and MPLC (30% → 50% MeOH in H2O), fraction F5 afforded 13 (135 mg), 3 (26 mg), 2 (21 mg), and 5 (272 mg). The retention times (tR) of new compounds 1–5 from analysis-type HPLC (20% → 80% MeOH in H2O over 8.0 min followed by 100% MeOH to 10 min, 1.0 ml/min, 20 ℃) were 7.8, 8.8, 7.9, 8.4, and 6.7 min, respectively.
Rauvotetraphylline A (1): yellowish, amorphous powder; [α]D16 +16.2 (c 0.20, MeOH); UV (MeOH) λmax: 211, 222 (sh), 275, 310 (sh) nm; IR (KBr) λmax 3407, 2923, 2892, 1630, 1596, 1454, 1382, 1364, 1236, 1190, 1144, 1114, 1059, 997, 798, 743 cm–1; 1H and 13C NMR data see Tables 1 and 2; ESIMS (pos.): m/z 343 [M + H]+; HRESIMS (pos.): m/z 343.2024 (calcd for C20H27N2O3, 343.2021).
Rauvotetraphylline B (2): yellowish, amorphous powder; [α]D16+52.7 (c 0.21, MeOH); UV (MeOH) λmax: 225, 265 (sh), 272, 282 (sh), 290 (sh) nm; IR (KBr) λmax 3406, 2921, 2856, 1608, 1568, 1452, 1411, 1384, 1335, 1238, 1170, 1075, 1026, 743 cm–1; 1H and 13C NMR data see Tables 1 and 2; ESIMS (pos.): m/z 548 [M + H]+; HRESIMS (pos.): m/z 548.2768 (calcd for C31H38N3O6, 548.2760).
Rauvotetraphylline C (3): yellowish, amorphous powder; [α]D16 +31.4 (c 0.19, MeOH); UV (MeOH) λmax: 225, 270, 284 (sh), 293 (sh) nm; IR (KBr) vmax 3405, 2918, 1669, 1622, 1572, 1452, 1419, 1384, 1362, 1335, 1259, 1238, 1170, 1075, 1025, 746 cm–1; 1H and 13C NMR data see Tables 1 and 2; ESIMS (pos.): m/z 511 [M + H]+; HRESIMS (pos.): m/z 511.2431 (calcd for C28H35N2O7, 511.2444).
Rauvotetraphylline D (4): yellowish, amorphous powder; [α]D14 +26.5 (c 0.20, CHCl3); UV (MeOH) λmax: 221, 226 (sh), 253 (sh) nm; IR (KBr) vmax 3431, 2964, 2931, 1741, 1695, 1673, 1620, 1592, 1468, 1453, 1363, 1231, 1177, 1138, 1034, 980, 952, 774, 753 cm–1; 1H and 13C NMR data see Tables 1 and 2; ESIMS (pos.): m/z 391 [M + H]+; HRESIMS (pos.): m/z 391.2019 (calcd for C24H27N2O3, 391.2021).
Rauvotetraphylline E (5): yellowish, amorphous powder; [α]D15 +136.8 (c 0.20, MeOH); UV (MeOH) λmax: 218 (sh), 234 (sh), 252, 260 (sh), 308, 370 nm; IR (KBr) vmax 3423, 3066, 2976, 2907, 1639, 1550, 1529, 1504, 1448, 1384, 1364, 1332, 1250, 1180, 1129, 790, 754 cm–1; 1H and 13C NMR data see Tables 1 and 2; ESIMS (pos.): m/z 335 [M + H]+; HRESIMS (pos.): m/z 335.1388 (calcd for C20H19N2O3, 335.1395).
Cytotoxicity Assay. The cytotoxicity assay was performed according to the MTT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide] method, 19 by use of the following five human cancer cell lines: human myeloid leukemia HL-60, hepatocellular carcinoma SMMC-7721, lung cancer A-549, breast cancer MCF-7, and colon cancer SW-480. The IC50 values were calculated by Reed and Muench's method.20
Notes
Electronic Supplementary Material
Supplementary material is available in the online version of this article at http://dx.doi.org/10.1007/s13659-012-0012-5 and is accessible for authorized users.
Acknowledgments
This work was financially supported by National Basic Research Program of China (973 Program) 2009CB522300, the "West Light" program of Chinese Academy of Sciences, and Natural Product Library Program of BioBioPha.
References
-
1.D. L. Bemis, J. L. Capodice, P. Gorroochurn, A. E. Katz, R. Buttyan, Int. J. Oncol. 29, 1065-1073 (2006) PubMed Google Scholar
-
2.C. W. Wright, J. D. Phillipson, S. O. Awe, G. C. Kirby, D. C. Warhurst, J. Quetin-Leclercq, L. Angenot, Phytother. Res. 10, 361-363 (1996) CrossRef PubMed Google Scholar
-
3.K. Hiwada, Ketsuatsu 13, 325-333 (2006) PubMed Google Scholar
-
4.Neuss, N. Indole and Biogenetically Related Alkaloids; Academic Press:New York, 1980; Chapter 17. PubMed Google Scholar
-
5.Y. Gao, F. Wang, D. S. Zhou, Y. Li, J. K. Liu, Nat. Prod. Bioprospect. 1, 104-107 (2011) CrossRef PubMed Google Scholar
-
6.P. Timmins, W. E. Court, Phytochemistry 15, 733-735 (1976) CrossRef PubMed Google Scholar
-
7.C. Kan, P. Potier, S. K. Kan, R. Jokela, M. Lounasma, Phytochemistry 25, 1783-1784 (1986) CrossRef PubMed Google Scholar
-
8.H. R. Arthur, S. R. Johns, J. A. Lamberton, S. N. Loo, Aust. J. Chem. 21, 1399-1401 (1968) CrossRef PubMed Google Scholar
-
9.J. A. Martìnez, H. Velez, T. Santana, Phytochemistry 28, 961-962 (1989) CrossRef PubMed Google Scholar
-
10.A. Itoh, T. Kumashiro, M. Yamaguchi, N. NagaKura, Y. Mizushina, T. Nishi, T. Tanahashi, J. Nat. Prod. 68, 848-852 (2005) CrossRef PubMed Google Scholar
-
11.R. Sheludko, I. Gerasimenko, H. Kolshorn, J. Stöckigt, J. Nat. Prod. 65, 1006-1010 (2002) CrossRef PubMed Google Scholar
-
12.H. Schübel, A. Treiber, J. Stökigt, Helv. Chim. Acta 67, 2078-2081 (1984) CrossRef PubMed Google Scholar
-
13.R. Verpoorte, J. Nat. Prod. 1, 1-25 (1986) PubMed Google Scholar
-
14.D. L. Boger, R.J. Wysocki Jr, J. Org. Chem. 54, 714-718 (1989) CrossRef PubMed Google Scholar
-
15.J. M. Yu, T. Wang, X. X. Liu, J. Deschamps, J. FlippenAnderson, X. B. Liao, J. M. Cook, J. Org. Chem. 68, 7565-7581 (2003) CrossRef PubMed Google Scholar
-
16.L. Katoa, R. M. Bragaa, I. Kochb, L. S. Kinoshitab, Phytochemistry 60, 315-320 (2002) CrossRef PubMed Google Scholar
-
17.L. Li, H. P. He, H. Zhou, X. Hao, J. Nat. Prod. Res. Dev. 19, 235-239 (2007) PubMed Google Scholar
-
18.J. L. Pousset, J. Poisson, L. Olivier, J. Le Men, M. M. Janot, C. R.Acad. Sci. 261, 5538 (1965) PubMed Google Scholar
-
19.T. Mosmann, J. Immunol. Methods 65, 55-63 (1983) CrossRef PubMed Google Scholar
-
20.L. J. Reed, H. Muench, Am. J. Hyg. 27, 493-497 (1938) PubMed Google Scholar
Copyright information
© The Author(s) 2012
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
