


Inhibition of P-glycoprotein by two artemisinin derivatives
Abstract
P-Glycoprotein/MDR1 represents an important component of the blood brain barrier and contributes to multidrug resistance. We investigated two derivatives of the anti-malarial artemisinin, SM616 and GHP-AJM-3/23, concerning their ability to interact with P-glycoprotein. The ability of the two compounds to inhibit P-glycoprotein(P-gp) activity was examined in sensitive CCRF-CEM and P-gp over-expressing and multidrug-resistant CEM/ADR5000 cells as well as in porcine brain capillary endothelial cells(PBCEC) by means of calcein-AM assays. Verapamil as well-known P-gp inhibitor was used as control drug. CEM/ADR5000 cells exhibited cross-resistance to GHP-AJM-3/23, but slight collateral sensitivity to SM616. Furthermore, SM616 inhibited calcein efflux both in CEM/ADR5000 and PBCEC, whereas GHP-AJM-3/23 did only increase calcein fluorescence in PBCEC, but not CEM/ADR5000. This may be explained by the fact that CEM/ADR5000 only express P-gp but not other ATP-binding cassette transporters, whereas PBCEC are known to express several ABC transporters and calcein is transported by more than one ABC transporter. Hence, SM616 may be the more specific P-gp inhibitor. In conclusion, the collateral sensitivity of SM616 as well as the inhibition of calcein efflux in both CEM/ADR5000 cells and PBCEC indicate that this compound may be a promising P-gp inhibitor to treat cancer therapy and to overcome the blood brain barrier.Keywords
blood brain barrier calcein multidrug resistance P-glycoproteinIntroduction
The medicinal herb, Artemisia annua L. (qinghao, Sweet Wormwood) is traditionally used in Chinese medicine against different ailments, such as fever, chills, nose bleeding, headache, jaundice, lupus erythematosus and hemorrhages. Its active principle is the 1, 2, 4-trioxane artemisinin, which was found to have anti-malarial activity in 1972.1 In the following years, several semisynthetic derivatives were developed to increase solubility in oil and water, i.e. artemether, arteether, artesunate. These substances are generally well-tolerated.2
In the 1990s, artemisinin and its derivatives attracted the attention of cancer researchers as their anti-proliferative activity was discovered.3-8 Artesunate was shown to inhibit tumor cell growth by cell cycle arrest9-12 and apoptosis induction in tumor cells.7, 13 Furthermore, it inhibits angiogenesis and metastasis in vitro and in vivo, 14-17 and induces oxidative DNA damage.18, 19
The activity of artemisinin derivatives in malaria parasites is mainly based on the formation of reactive oxygen species (ROS) and carbon-centered radicals. While both damage different target proteins, ROS can also lead to lipid peroxidation. The activity against cancer cells seems to follow a similar pathway, as the over-expression of anti-oxidant genes decreases cytotoxicity of artesunate.9, 20-22 Furthermore, iron (Ⅱ) glycine sulfate (Ferrosanol®) and transferrin were shown to increase cytotoxicity of artesunate, 23, 24 pointing to a role of iron ions in a Fenton-type reaction of artemisinins.
The epidermal growth factor receptor (EGFR) is linked to resistance against artesunate. Increased EGFR expression correlated with decreased artesunate toxicity in a COMPARE analysis of IC50 values of artesunate and genome-wide mRNA expression in the NCI cell line panel.9 Upon combination of artesunate with the EGFR tyrosine kinase inhibitor, erlotinib, supra-additive effects were observed in EGFR over-expressing cells.25 Furthermore, artesunate affects kinases of signalling pathways downstream of EGFR.26 Resistance to artesunate is also mediated by NF-κB.27
A remarkable feature is that multidrug-resistant Plasmodium strains as well as multidrug-resistant cancer cells do not exhibit cross-resistance to artesunate.9, 28-30 The phenomenon of multidrug resistance is often caused by over-expression of ATP-binding cassette (ABC) transporters resulting in an increased efflux of their substrates.30-32 Over-expression of the ABC-transporter P-glycoprotein (P-gp)/MDRI result in resistance to a wide variety of cancer drugs, e.g. anthracyclines, Vinca alkaloids, epipodophyllo-toxins, taxanes and others. P-gp is constitutively found in the blood-brain barrier, where it functions as an efflux pump to prevent xenobiotics from passing through the barrier. Artesunate was tested in several multidrug-resistant and drug-sensitive cell lines9, 29, 30 and no cross-resistance was observed. There is, however, not only a need for non-cross-resistant drugs, but also for inhibitors of P-gp function to improve standard chemotherapy of tumors and to overcome the blood brain barrier for pharmacological intervention in neurological diseases.
In the present investigation, two new artemisinin derivatives have been tested, SM616 and GHP-AJM-3/23 in sensitive CCRF-/CEM and P-gp over-expressing and multidrug resistant CEM/ADR5000 leukemia cells as well as in porcine brain capillary endothelial cells (PBCEC). The capability of the artemisinin derivatives to modulate the efflux function of P-gp was measured by means of a calcein-AM assay.33 The results of the compounds were compared to the activity of verapamil, a well-described modulator of P-gp mediated efflux.
Results and Discussion
Growth Inhibition Assay. First, the cytotoxicity of the two artemisinin derivatives was determined in CCRF-CEM and CEM/ADR5000 leukemia cells. The IC50 values are shown in Table 1. The P-gp over-expressung and multidrug-resistant CEM/ADR5000 cells were 11.1-fold more resistant to GHPAJM-3/23 than the sensitive CCRF-CEM cells. The opposite was observed for SM616: CEM/ADR5000 cells were more sensitive to the compound than the parental CCRF-CEM cell line. The degree of resistance in CEM/ADR5000 cells was 0.61. As a control, the response of the leukemia cells to doxorubicin was investigated. CEM/ADR5000 cells revealed 4260-fold resistance towards doxorubicin.
50% inhibition concentration (IC50) and relative degrees of resistance of two artemisinin derivatives and doxorubicin as control drug in human leukemic CCRF-CEM and CEM/ADR5000 cells determined by growth inhibition assay
Calcein-AM Assay with Adherent PBCEC. The efflux of calcein from PBCEC was measured to determine the effects of the two artemisinin derivatives on the activity of P-gp. Figure 2 shows the averaged results of each two experiments. The intracellular calcein fluorescence considerable increased after treatment with GHP-AJM-3/23. A strong increase was observed within the first minutes of incubation. Although the intracellular accumulation slightly decreased afterwards, the level of untreated control cells was not reached (Figure 2a). The intracellular calcein fluorescence was stable upon application of SM616 and rather increased after 150 min, which speaks for an efficient inhibition of P-gp-mediated calcein efflux (Figure 2b).
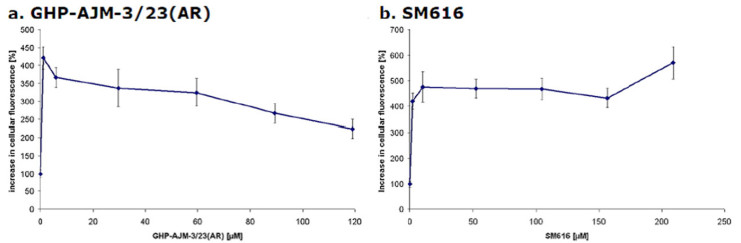
Inhibition of calcein efflux by (a) GHP-AJM-3/23 and (b) SM616 in PBCEC
In each experiment, three different control measurements were carried out. (1) Krebs-Ringer-Buffer was added instead of calcein-AM to monitor the autofluorescence of PBCEC. The mean fluorescence value was subsequently subtracted from all other measured values. (2) The uptake and efflux of calcein as measured by adding Krebs-Ringer-Buffer instead of artemisinins. (3) As positive control, 25 μM Verapamil was used. Verapamil increased the cellular calcein fluorescence by 550 to 600%.
Calcein-AM Assay with CCRF-CEM and CEM/ADR5000 Leukemia Cells in Suspension. As a next step, the calcein fluorescence was studied in leukemia cell lines. The measurement of intracellular fluorescence was performed with flow cytometry, since the leukemia cells grew in suspension culture. Using forward sideward scatter, cell debris and cell doublettes were excluded. The suspensions were treated with propidium iodide to exclude dead cells from the measurement. Samples were measured and analyzed with the software CellQuest Pro.
The flow cytometric histograms for the intracellular calcein fluorescence with and without treatment of cells with SM616 are shown in Figure 3. SM616 had no effect in parental CCRF-CEM cells without P-gp expression (Figure 3a). In P-gp overexpressing CEM/ADR5000 cells, however, SM616 caused a considerable dose-dependent increase of calcein fluorescence, indicating inhibition of P-gp-mediated efflux (Figure 3b). When the same experiment was performed with GHP-AJM-3/23, increased calcein fluorescence was not observed in CEM/ADR5000 cells, indicating that this compound was not able to block P-gp function (Figure 4). As a control drug, we used verapamil, which is a well-known P-hp inhibitor. The histograms in Figure 5 demonstrate, that a verapamil-induced increase of calcein fluorescence was measured in CEM/ADR5000 cells (Figure 5a), but not in CCRF-CEM cells (Figure 5b).
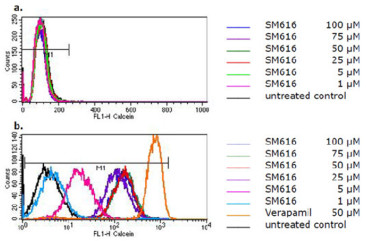
Flow cytometric histograms of intracellular calcein fluorescence in (a) CCRF-CEM and (b) CEM/ADR5000 cells after treatment with various concentrations of SM616
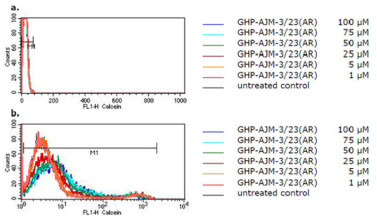
Flow cytometric histograms of intracellular calcein fluorescence in (a) CCRF-CEM and (b) CEM/ADR5000 cells after treatment with various concentrations of GHP-AJM-3/23(AR)
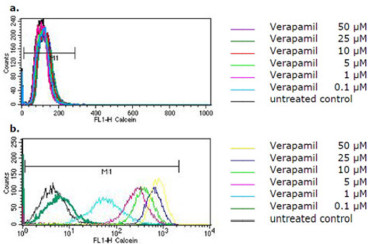
Flow cytometric histograms of intracellular calcein fluorescence in (a) CCRF-CEM and (b) CEM/ADR5000 cells after treatment with various concentrations of verapamil
Multidrug-resistant CEM/ADR5000 cells were crossresistant to GHP-AJM-3/23 but not SM616. P-glycoprotein is a broad spectrum transporter for a wide diversity of compounds. It is a characteristic feature of P-gp that it translocates a wide variety of chemically diverse compounds. While many speculations on the mechanism of action of P-glycoprotein have been made, the only common property of P-glycoprotein substrates is their relative hydrophobic, amphiphilic nature.36, 37
Interestingly, CEM/ADR5000 cells did not reveal crossresistance to SM616. This phenomenon is described as 'collateral sensitivity' and was first observed in the reaction of bacteria towards different antibiotics.38 It is also well-known in P-gp expressing and otherwise multidrug-resistant cells.39 Rapid ATP-consumption by futile cycling, increased ROS production and glutathione depletion, altered lipophilicity of cell membranes and other mechanisms were discussed as possible mechanisms to explain collateral sensitivity. The reasons are unknown yet, why CEM/ADR5000 cells exhibited collateral sensitivity towards SM616.
Although both compounds are derivatives of artemisinin, they are structurally very different. GHP-AJM-3/23 is a dimer molecule of artemisinin, while SM616 is a monomer with an additional side chain. CEM/ADR5000 cells were crossresistant to one compound, but collateral sensitive to the other one. Furthermore, the calcein efflux was inhibited by SM616 but not by GHP-AJM-3/23. These data may speak for different binding sites of the two compounds at P-gp.
The binding of small molecules to P-gp is a matter of a long and inconclusive discussion. While initially one binding domain and subsequently two binding sites have been proposed for P-glycoprotein, 40 more recent investigations suggested multiple different binding sites.41, 42 An alternative model hypothesized that P-glycoprotein extrude diverse drugs by an induced-fit mechanism.43 Recently, homology models for P-glycoprotein based on the crystal structure of the bacterial ABC transporter from Staphylococcus aureus Sav1866 have been described.44 Site-directed mutagenesis experiments fit to these putative binding sites of the homology models, 45 but a final proof can only be delivered by drug-protein crystal structures. In own investigations, we also found an unexpected broad diversity of natural products, including several artemisinin derivatives as substrates or inhibitors of P-gp.46, 16 Considering this complex and unresolved situation, the substrate specificity of P-glycoprotein may be much broader than estimated thus far.
Whereas SM616 inhibited calcein efflux both in CEM/ADR5000 cells in PBCEC, GHP-AJM-3/23(AR) inhibited calcein efflux in PBCEC but not in CEM/ADR5000 cells. This result may be explained by the fact that other ABC transporters in addition to P-gp are also expressed in PBCEC.47 MRP1 mRNA was found in cultured PBCEC, although to a 10-fold lower level than PGP mRNA. As calceinAM is also a substrate for MRP1, 48 higher intracellular calcein content could be explained by interaction of GHP-AJM-3/23(AR) with MRP1. By contrast, CEM/ADR5000 have been shown to express P-gp but not other ABC transporters.35
In conclusion, two artemisinin derivatives, SM616 and GHP-AJM-3/23 showed different behavior in P-gp expressing cells. The missing cross-resistance of SM616 as well as the inhibition of calcein efflux in both CEM/ADR5000 cells and PBCEC indicate that this compound may be a promising P-gp inhibitor to treat cancer therapy and overcome the blood brain barrier. As artemisinins were generally well-tolerated in clinical treatment against malaria in the past, SM616 might provide a non-toxic chemosensitizer. This hypothesis warrants more detailed investigations in the future.
Experimental Section
Drugs. The artemisinin derivative, SM616 ((16S)-cyano-(pbromo-phenyl)-methyl-12β-deoxoartemisinyl ether), was synthesized by Dr. Ying Li (Department of Synthetic Chemistry, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China). The compound is solid at room temperature and has a molecular weight of 478.38 g/mol. Stock solutions were prepared in DMSO. GHP-AJM-3/23 was synthesized by Dr. Gary H. Posner (Department of Chemistry, Johns Hopkins University, Baltimore MD, USA). It is an artemisinin-dimer molecule, which is solid at room temperature. The molecular weight is 838.96 g/mol. Stock solutions were prepared in DMSO. (±)-Verapamil hydrochloride was obtained from Sigma-Aldrich (Deisendorf, Germany) and calcein-acetoxymethylester (calcein-AM) was obtained from Invitrogen (Karlsruhe, Germany).
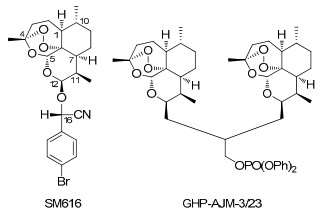
Chemical structures of SM616 and GHP-AJM-3/23
Cells. The human leukemia cell lines CCRF-CEM and CEM/ADR5000 were obtained from Dr. Axel Sauerbrey (Department of Pediatrics, University of Jena, Germany). Cells were cultivated in RPMI 1640 medium, 2.0 g/L NaHCO3 with stable L-glutamine supplemented with 10% fetal calf serum and 0.1 U/ml/0.1 mg/ml Penicillin/Streptomycin (all Biochrom, Berlin, Germany) in a humidified 5% CO2 atmosphere at 37 ℃. CEM/ADR5000 cells were incubated in RPMI 1640 medium containing 5000 ng/ml Doxorubicin for 24 h once a week. The CEM/ADR5000 line was developed by Kimmig et al. (1990).34 The cells express specifically P-gp/MDRI, but none of the other members of the ABC transporter family.35
Porcine brain capillary endothelial cells (PBCEC) were isolated following a procedure previously described.33 Brains of freshly slaughtered animals were obtained from a local slaughterhouse and transported and stored in artificial cerebrospinal fluid (aCSF). The cortical grey matter was isolated by removing meninges, choroid plexus and blood vessels and minced to pieces of 1 to 2 mm3. The mass was then incubated in culture medium (1) containing 0.5% dispase Ⅱ (Hoffmann LaRoche, Mannheim, Germany) for 2 h at 37 ℃. The homogenate was centrifuged at 1000 × g for 10 min. After removing the supernatant, the pellet was suspended in culture medium containing 15% dextran (Sigma, Taufkirchen, Germany). In the following centrifugation at 5800 g for 10 min, the brain tissue was separated from the microvessels which form the pellet. Subsequently, the capillaries were incubated in culture medium containing 1 mg/mL collagenase-dispase (Hoffmann LaRoche, Mannheim, Germany) for 1.5 to 2 h. By filtration through a 150-μm Polymon® mesh (NeoLab Migge, Heidelberg, Germany) and another centrifugation step at 130 g for 10 min, the cell suspension was further purified. Subsequently, cells were held in culture medium containing 10% horse serum. The suspension was then added to a discontinuous Percoll® (Pharmacia, Freiburg, Germany) gradient, consisting of Percoll® 1.03 g/mL and 1.07 g/mL at a ratio of 4 : 3. It is critical that the following centrifugation step at 1000 × g for 10 min is performed without breaking to prevent an unwanted mixture of the two phases. While debris was collected in the low-density fraction and erythrocytes in the high-density fraction, the capillary endothelial cells remain at the interface, where they were collected. Two more centrifugation steps at 130 × g at 10 min followed. The cells were then suspended in culture medium containing horse serum. Cells were seeded at a density of 250, 000 cells/cm2 in 96-well-plates and cultivated in a humidified 5% CO2 atmosphere at 37 ℃. The culture medium was medium 199, supplemented with 0.8 mM Lglutamine, penicillin/streptomycin (100 U/mL; 100 μg/mL), 100 μg/mL kanamycin and 10 mM HEPES, pH 7.4 (all from Biochrom, Berlin, Germany).
Growth Inhibition Assay. CEM-cells were seeded at a density of 50, 000 cells/ml in RPMI medium containing 10% fetal calf serum. Drug solutions were immediately added in varying concentrations. After seven days, cells were counted using a Neubauer counting chamber. Cell numbers were counted 16 times and the results were averaged. The resulting growth curves represent the net outcome of cell proliferation and cell death. The substance concentration resulting in 50% growth inhibition (IC50 value) was calculated from the dose-response curves, assuming linearity of the curve in the proximity of the IC50. The following formula was used:
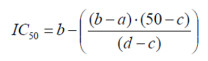
|
a = concentration of the substance resulting in less than 50% growth inhibition
b = concentration of the substance resulting in more than 50% growth inhibition
c = measured cell growth (% of control) at concentration b
d = measured cell growth (% of control) at concentration a
Calcein-AM Assay with Aherent PBCEC. Cells were isolated and cultivated in collagen-coated 96-well plates as described above. Medium was changed on day after preparation and from there on at every second day. Six days after isolation, medium was changed and the cells were held in medium lacking horse serum. On the seventh day after preparation, the assay was performed, provided that a stable monolayer had developed. Substance stock solutions were prepared in DMSO. Dilutions were made in aqua bidest. assured that the final DMSO concentration on the cells did not exceed 1%. In the assay, medium was removed and cells were washed twice with Krebs-Ringer-buffer (KRB). The composition of KRB was: 142 mM NaCl, 3 mM KCl, 1.8 mM K2HPO4 × 3 H2O, 10 mM HEPES, 4 mM D-Glucose, 1.3 mM MgCl2 × 6 H2O, 1.4 mM CaCl2 × 2 H2O. Substance solutions in different concentrations were added and the plates were incubated for 15 min at 37 ℃. An equal volume of 2 μM calcein-acetoxymethylester (calcein-AM) was added (resulting in 1 μM calcein-AMsolution in the assay) and incubated for 30 min at 37 ℃. The layer was then washed twice with KRB (cooled down to 4 ℃) to stop calcein-AM hydrolysis and remove extracellular calcein. For cell lysis, plates were incubated at 37 ℃ in 1% TritonX-100 for 20 min. Afterwards, fluorescence was measured with Fluoroskan Ascent plate reader (Labsystems, Waltham MA, USA) at 485 nm excitation and 520 nm emission.
Calcein-AM Assay with CCRF-CEM and CEM/ADR5000 Leukemia Cells in Suspension. Cells were cultivated in RPMI 1640 medium as described above. Substance stock solutions were prepared in DMSO. Dilutions were made in aqua bidest. The final DMSO concentration on the cells did not exceed 1%. Cells were seeded at a density of 2.5 ×106 cells/ml in RPMI 1640 medium. An aliquot of the substance solution was added and incubated at 37 ℃ for 15 min. Subsequently, the same volume of 4 μM Calcein-AM-solution was added, resulting in a final concentration of 1 μM CalceinAM solution. The suspension was incubated at 37 ℃ for 30 min. To stop the reaction, the suspension was cooled to 4℃ and cells were centrifuged at 500 × g for 5 min. The pellet was washed twice with 4 ℃ cold RPMI 1640 medium and was finally suspended therein. To each sample, propidium iodide was added to a final concentration of 0.5 μM. Afterwards, fluorescence was measured with FACSCalibur up to 20, 000 counts, according to the manufacturer's instructions (Becton and Dickinson Biosciences, San José, USA). The excitating laser-light had a wavelength of 488 nm. Calcein was measured with FL1-channel, which measures light that passed through a 530/30 bandpass filter. Propidium iodide was measured with FL3-channel, measuring light passing through a 650 nm longpass filter. Data was analysed using the CellQuest Pro software (Becton and Dickinson Biosciences, San José, USA).
References
-
1.Y. Li, Y. L. Wu, Curr. Med. Chem. 10, 2197-2230 (2003) CrossRef PubMed Google Scholar
-
2.T. Efferth, B. Kaina, Crit. Rev. Toxicol. 40, 405-421 (2010) CrossRef PubMed Google Scholar
-
3.W. S. Sun, J. X. Han, W. Y. Yang, D. A. Deng, X. F. Yue, Acta Pharmacol. Sin. 13, 541-543 (1992) PubMed Google Scholar
-
4.H. J. Woerdenbag, T. A. Moskal, N. Pras, T. M. Malingre, F. S. el-Feraly, H. H. Kampinga, A. W. Konings, J. Nat. Prod. 56, 845-849 (1993) PubMed Google Scholar
-
5.G. Q. Zheng, Planta Med. 60, 54-57 (1994) CrossRef PubMed Google Scholar
-
6.A. C. Beekman, H. J. Woerdenbag, Uden W. van, N. Pras, A. W. Konings, H. V. Wikstrom, J. Pharm. Pharmacol. 49, 1254-1258 (1997) CrossRef PubMed Google Scholar
-
7.T. Efferth, G. Rücker, M. Falkenberg, D. Manns, A. Olbrich, U. Fabry, R. Osieka, Arzneimittelforschung 46, 196-200 (1996) PubMed Google Scholar
-
8.T. Efferth, H. Dunstan, A. Sauerbrey, H. Miyachi, C. R. Chitambar, Int. J. Oncol. 18, 767-773 (2001) PubMed Google Scholar
-
9.T. Efferth, A. Sauerbrey, A. Olbrich, E. Gebhart, P. Rauch, H. O. Weber, J. G. Hengstler, M. E. Halatsch, M. Volm, K. D. Tew, D. D. Ross, J. O. Funk, Mol. Pharmacol. 64, 382-394 (2003) CrossRef PubMed Google Scholar
-
10.L. Steinbrück, G. Pereira, T. Efferth, Cancer Genomics Proteomics 7, 337-346 (2010) PubMed Google Scholar
-
11.S. Sertel, T. Eichhorn, S. Sieber, A. Sauer, J. Weiss, P. K. Plinkert, T. Efferth, Chem. Biol. Interact. 185, 42-52 (2010a) CrossRef PubMed Google Scholar
-
12.S. Sertel, T. Eichhorn, C. H. Simon, P. K. Plinkert, S. W. Johnson, T. Efferth, Molecules 15, 2886-2910 (2010b) CrossRef PubMed Google Scholar
-
13.T. Efferth, M. Giaisi, A. Merling, P. H. Krammer, M. Li-Weber, PLoS One 2, e693 (2007) PubMed Google Scholar
-
14.R. Dell'Eva, U. Pfeffer, R. Vené, L. Anfosso, A. Forlani, A. Albini, T. Efferth, Biochem. Pharmacol. 68, 2359-2366 (2004) CrossRef PubMed Google Scholar
-
15.L. Anfosso, T. Efferth, A. Albini, U. Pfeffer, Pharmacogenomics J. 6, 269-278 (2006) CrossRef PubMed Google Scholar
-
16.S. Soomro, T. Langenberg, A. Mahringer, V. B. Konkimalla, C. Horwedel, P. Holenya, A. Brand, C. Cetin, G. Fricker, M. Dewerchin, P. Carmeliet, E. M. Conway, H. Jansen, T. J. Efferth, Cell Mol. Med. 15, 1122-1135 (2011) CrossRef PubMed Google Scholar
-
17.S. A. Rasheed, T. Efferth, I. A. Asangani, H. Allgayer, Int. J. Cancer 127, 1475-1485 (2010) CrossRef PubMed Google Scholar
-
18.P. C. Li, E. Lam, W. P. Roos, M. Z. Zdzienicka, B. Kaina, T. Efferth, Cancer Res. 68, 4347-4351 (2008) CrossRef PubMed Google Scholar
-
19.Berdelle, N.; Nikolova, T.; Quiros, S.; Efferth, T.; Kaina, B. Mol. Cancer Ther. 2011[Epub ahead of print]. PubMed Google Scholar
-
20.T. Efferth, M. M. Briehl, M. E. Tome, Int. J. Oncol. 23, 1231-1235 (2003) PubMed Google Scholar
-
21.T Efferth, F. Oesch, Biochem. Pharmacol. 68, 3-10 (2004) CrossRef PubMed Google Scholar
-
22.T. Efferth, M. Volm, In Vivo 19, 225-232 (2005) PubMed Google Scholar
-
23.T. Efferth, A. Benakis, M. R. Romero, M. Tomicic, R. Rauh, D. Steinbach, R. Häfer, T. Stamminger, F. Oesch, B. Kaina, M. Marschall, Free Radic. Biol. Med. 37, 998-1009 (2004) CrossRef PubMed Google Scholar
-
24.G. Kelter, D. Steinbach, V. B. Konkimalla, T. Tahara, S. Taketani, H. H. Fiebig, T. Efferth, PLoS One 2, e798 (2007) PubMed Google Scholar
-
25.T. Efferth, T. Ramirez, E. Gebhart, M. E. Halatsch, Biochem. Pharmacol. 67, 1689-1700 (2004) CrossRef PubMed Google Scholar
-
26.V. B. Konkimalla, J. A. McCubrey, T. Efferth, Curr. Cancer Drug Targets 9, 72-80 (2009) CrossRef PubMed Google Scholar
-
27.B. Bachmeier, I. Fichtner, P. H. Killian, E. Kronski, U. Pfeffer, T. Efferth, PLoS One 6, e20550 (2011) PubMed Google Scholar
-
28.R. Price, M. van Vugt, F. Nosten, C. Luxemburger, A. Brockman, L. Phaipun, T. Chongsuphajaisiddhi, N. White, Am. J. Trop. Med. Hyg. 59, 883-888 (1998) CrossRef PubMed Google Scholar
-
29.T. Efferth, M. Davey, A. Olbrich, G. Rücker, E. Gebhart, R. Davey, Blood Cells Mol. Dis. 28, 160-168 (2002) CrossRef PubMed Google Scholar
-
30.T. Efferth, Curr. Mol. Med. 1, 45-65 (2001) CrossRef PubMed Google Scholar
-
31.J. P. Gillet, T. Efferth, J. Remacle, Biochim. Biophys. Acta 1775, 237-262 (2007) PubMed Google Scholar
-
32.Eichhorn, T.; Efferth, T. J. Ethnopharmacol. 2011[Epub ahead of print]. PubMed Google Scholar
-
33.B. Bauer, D. S. Miller, G. Fricker, Pharm. Res. 20, 1170-1176 (2003) CrossRef PubMed Google Scholar
-
34.A. Kimmig, V. Gekeler, M. Neumann, G. Frese, R. Handgretinger, G. Kardos, H. Diddens, D. Niethammer, Cancer Res. 50, 6793-6799 (1990) PubMed Google Scholar
-
35.J. P. Gillet, T. Efferth, D. Steinbach, J. Hamels, F. de Longueville, V. Bertholet, J. Remacle, Cancer Res. 64, 8987-8993 (2004) CrossRef PubMed Google Scholar
-
36.M. M. Gottesman, I. Pastan, Annu. Rev. Biochem. 62, 385-427 (1993) CrossRef PubMed Google Scholar
-
37.J. Rautio, J. E. Humphreys, L. O. Webster, Drug Metab. Dispos. 34, 786-792 (2006) CrossRef PubMed Google Scholar
-
38.W. Szybalski, V. Bryson, J. Bacteriol. 64, 489-499 (1952) PubMed Google Scholar
-
39.M. D. Hall, M. D. Handley, M. M. Gottesman, Trends Pharmacol. Sci. 30, 546-556 (2009) CrossRef PubMed Google Scholar
-
40.A. H. Schinkel, E. Wagenaar, C. A. Mol, L. van Deemter, J. Clin. Invest. 97, 2517-2524 (1996) CrossRef PubMed Google Scholar
-
41.S. Ayesh, Y. M. Shao, W. D. Stein, Biochim. Biophys. Acta 1316, 8-18 (1996) CrossRef PubMed Google Scholar
-
42.M. J. Borgnia, G. D. Eytan, Y. G. Assaraf, J. Biol. Chem. 271, 3163-3171 (1996) CrossRef PubMed Google Scholar
-
43.A. R. Safa, Curr. Med. Chem. Anticancer Agents 4, 1-17 (2004) CrossRef PubMed Google Scholar
-
44.C. Globisch, I. K. Pajeva, M. Wiese, Chem. Med. Chem. 3, 280-295 (2008) CrossRef PubMed Google Scholar
-
45.A. B. Shapiro, V. Ling, Eur. J. Biochem. 250, 122-129 (1997) CrossRef PubMed Google Scholar
-
46.A. Mahringer, S. Karamustafa, D. Klotz, S. Kahl, V. B. Konkimalla, Y. Wang, J. Wang, H. Y. Liu, H. Boechzelt, X. Hao, R. Bauer, G. Fricker, T. Efferth, Cancer Genomics Proteomics 7, 191-205 (2010) PubMed Google Scholar
-
47.M. Török, J. Huwyler, H. Gutmann, G. Fricker, J. Drewe, Exp. Brain Res. 153, 356-365 (2003) CrossRef PubMed Google Scholar
-
48.M. Stark, L. Rothem, G. Jansen, G. L. Scheffer, I. D. Goldman, Y. G. Assaraf, Mol. Pharmacol. 64, 220-227 (2003) CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2012
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
