


Novel indole and quinoline alkaloids from Melodinus yunnanensis
Abstract
6/7-Seco rearranged spiro-indolone alkaloids, meloyunines A(1) and B(2) and a monoterpenoid quinoline alkaloid meloyunine C(3) together with its possible intermediate 14, 15-dehydromelohenine B(4), and their precursor Δ14-vincamenine(5) were isolated from Melodinus yunnanensis. All structures were elucidated based on NMR, FTIR, UV, and MS spectroscopic data. The isolation of monoterpenoid indole, quinoline, and its immediate from the same plant chemically supported the biosynthesis of quinoline from indole. Compound 2 was cytotoxic against several human cancer cell linesKeywords
spiro-indolone quinoline meloyunine Melodinus yunnanensisIntroduction
Monoterpenoid indole alkaloids originate from the condensation of tryptophan with secologanin to produce strictosidine, which further alters by rearrangement to yield a dozen subgroups.1 Some of the remarkable quinoline alkaloids, such as quinine and camptothecin which are well known for their antimalarial and anticancer properties, respectively, have been proposed to arise by rearrangement of monoterpenoid indole alkaloids. In a possible route for quinine biosynthesis, the cleavage of a N1-C2 bond in the indole heterocyclic ring could generate new amine and keto functions. A new quinoline heterocycle would then be formed by combining this N-1 amine with a C-5 aldehyde produced by a tryptamine sidechain cleavage, producing cinchonidinone.2 Unlike quinine, the proposed biosynthesis of camptothecin includes a C2–C7 double bond oxidation to yield two carbonyls and an aldoltype condensation between C2 and C6 to form a quinoline ring. An in vivo tracer experiment has supported the prediction that the quinoline moiety originates from tryptophan.3 Also, melodinine B, a possible key intermediate of indole to quinoline alkaloids, has been reported.4 Plants of the genus Melodinus have been shown to be good sources of monoterpenoid indole and quinoline alkaloids.5 During our search for novel and bioactive monoterpenoid indole alkaloids from the family Apocynaceae, some representative skeletons and cytotoxic compounds were reported from the genera Alstonia6 and Melodinus.4,7 This paper describes skeletons and cytotoxic compounds were reported from the genera Alstonia6 and Melodinus.4,7 This paper describes the isolation, structural determination, proposed biosynthesis, and cytotoxic activities of 4 novel alkaloids (1–4) from M. yunnanensis.
Results and Discussion
Compound 1 was found to possess a molecular formula of C19H20N2O, as evidenced by high resolution electron spray ionization mass spectra (HRESIMS) at m/z 293.1650, in combination with 1H, 13C NMR, DEPT spectra, and appropriate for 11 degrees of unsaturation. The UV spectrum showed the presence of conjugated groups by showing maximum absorptions at 247 and 278 nm, and the IR spectrum indicated the presence of carbonyl and olefin groups (absorption bands at 1702 and 1610 cm-1, respectively). In the 1H NMR spectrum, two doublet (δH 7.55 and 7.13) and two triplet (δH 7.56 and 6.91) signals indicated that 1 was an unsubstituted indole alkaloid. In addition, signals for double bonds (δH 6.69 (d, J = 7.0 Hz, H-16), 5.42 (dd, J = 7.0, 1.4 Hz, H-17), 5.81 (m, H-14), and 5.64 (dd, J = 10.0, 1.4 Hz, H-15)), a methyl group (δH 0.62, t, H-18), and a methylene (δH 0.95 and 0.87, each 1H, m, H-19) were similar to those of ∆14-vincamenine (5).8 The 13C NMR and DEPT spectra of 1 showed signals of a methyl group (δC 8.5, q), four sp3 methylenes (δC 52.1, 53.1, 39.5, and 33.0), eight sp2 (δC 133.5, 126.7, 126.0, 125.0, 124.1, 120.3, 137.9, and 112.0) and one sp3 methines (δC 72.1), and three sp2 (δC 204.0, 159.8, and 120.8) and two sp3 quaternary carbons (δC 73.9 and 42.2). In comparison with those of 5, the four quaternary carbon signals at δC 131.0 (C-2), 134.8 (C-13), 129.2 (C-8), and 107.9 (C-7) of 5 were absent in 1, instead δC 204.0 (s), 73.9 (s), 159.8 (s), and 120.8 (s) were present in 1. In the heteronuclear multiple bond coherence (HMBC) spectrum of 1 (Figure 2), the correlation of δH 7.55 (1H, H-9) with δC 204.0 (s) suggested an indolone fragment, and the correlation of δH 6.69 (H-16), 1.88, 2.08 (H-6), and 3.10 (H-5) with δC 73.9 (s) led to the assignment of C-2 to a conjunct carbon of a spiral ring (Figure 1). Nuclear Overhauser Effect (NOE) correlation between H-21 and H-19 in the ROESY spectrum of 1 indicated three protons on the same side, a α-orientation identical to the configuration of its biosynthetic precursor, ∆14-vincamenine.9 Thus, the C-5 and 6 of the spiral ring (C) were on the upside of the planar indole configuration in a molecular model. Rings A, D, and E of 1 were same as those of 5, as indicated by the HMBC and ROESY spectra.
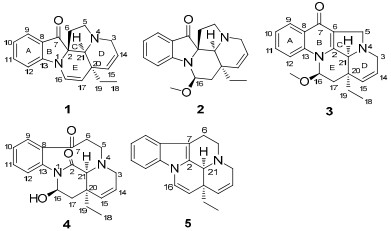
Structures of compounds 1-5.
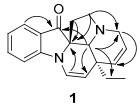
Key HMBC correlations of compound 1.
Compound 2 possessed a molecular formula C20H24N2O2 according to HRESIMS (m/z 325.1913, [M + H]+). The 1H and 13C NMR spectra were very similar to those of 1, except for δC 85.5 (d), 36.2 (t), and an additional methoxyl δC 54.2 (q) in 2 instead of the olefinic signals at C-16/17 in 1. This suggested an added methoxyl group at C-16 in 2, which was further supported by the HMBC spectrum, with correlations of δH 5.04 (1H, t, J = 8.0 Hz, H-16) with C-2, 13, and 17. In the ROESY spectrum, NOE correlation of δH 5.04 (H-16) with δH 0.77 and 0.87 (2H, m, H-19), and of δH 3.39 (3H, s, OCH3) with 2.19 (2H, m, H-6) placed the methoxyl as β. Other structural parts of 2 were identical to 1, as indicated by the HMBC and ROESY spectra.
Compound 3 possessed a molecular formula of C20H22N2O2, indicated by HRESIMS at m/z 323.1752 [M + H]+ in combination with 1H, 13C NMR, and DEPT spectra. Its UV spectrum showed a conjugation pattern (absorptions at 230 and 292 nm) different from 1. In the 1H NMR spectrum, four downfield protons (δH 8.34 (d, J = 8.4 Hz, H-9), 7.93 (d, J = 8.4 Hz, H-12), 7.68 (t, J = 8.4 Hz, H-11), and 7.37 (t, J = 8.4 Hz, H-10)) suggested a nonsubstituted quinolone rather than indole,10 further supported by the HMBC correlation of δH 8.34 (d, H-9) with a conjugated ketone signal at δC 176.0 (s, C-7). Detailed comparison of the NMR data of 2 and 3, indicated fused 6/5 rings (B and C) in 3 instead of corresponding spiral 5/5 rings in 2. Moreover, a methoxyl in β-orientation was supported by the NOE correlation of H-16 with H-19 in its ROESY spectrum.
The HRESIMS (m/z 327.1708, [M + H]+) of 4 established the molecular formula of C19H22N2O3. The NMR pattern was similar to melohenine B,4 except for olefinic signals [δC 122.7 (d) and 133.4 (d) and δH 5.83 (1H, m) and 5.74 (1H, d, J = 10.0 Hz)] observed in 4. The double bond was assumed to be at C-14/15 from comparison of spectral data from the two compounds and was further supported by the HMBC spectrum of 4, with correlations of δC 39.6 (s, C-20) with δH 5.83 (1H, m, H-14) and 5.74 (1H, d, H-15). NOE correlations of H-21 with H-19 and H-16 supported a 16β-OH for 4, identical to melohenine B.
The possible biosynthetic relationships of these new compounds in which they were derived from a common precursor 5 was proposed here (Figure 3). Different oxidation processes may have produced two kinds of intermediates, from which 4 was isolated. Further rearrangement then formed two new skeletons, including the spiro-indolone alkaloids (1 and 2), and a quinolone alkaloid (3). To our knowledge, this is the first report of the co-occurrence of monoterpenoid indoles (1, 2, and 5), a quinoline (3), and their key intermediates (4) in same plant, supporting the biosynthesis of quinoline from indole and concurring with previous in vivo tracer experiments in the literature.

Proposed biogenetic relationships of 1-5.
Compounds 1–5 were evaluated for their cytotoxicity against five human cancer cell lines, MCF-7 breast, SMMC-7721 hepatocellular carcinoma, HL-60 myeloid leukemia, SW480 colon cancer, and A-549 lung cancer. Compound 2 showed cytotoxicity against all cell lines, with IC50 values of 14.24, 19.08, 15.48, 13.29, and 40.0 μm, respectively, while the cisplatin control showed IC50 values of 21.90, 15.24, 1.05, 19.92, and 9.40 μm. The other compounds were inactive (IC50 > 40 μm).
Experimental Section
General Experimental Procedures. Optical rotations were measured with a Jasco P-1020 spectropolarimeter. UV spectra were recorded on a Shimadzu double-beam 210A spectrophotometer. IR (KBr) spectra were obtained on Bruker Tensor 27 infrared spectrophotometer. 1H, 13C and 2D NMR spectra were recorded on a Bruker avance Ⅲ-600 and AV-400 MHz NMR spectrometer with TMS as internal standard. MS data were obtained on API Qstar Pulsar Ⅰ spectrometer. C18
silica gel (20–45 μm) was bought from Fuji Chemical Ltd., Japan. MPLC was employed Büchi pumps system coupled with glass column (15 × 230 and 26 × 460 mm, respectively, C18 silica gel). HPLC was performed using Waters 600 pumps coupled with analytical and semipreparative Sunfire C18 columns (4.6 × 150 and 19 × 150 mm, respectively). The HPLC system employed a Waters 2996 photodiode array detector and a Waters fraction collector Ⅱ.
Plant Materials. Leaves and twigs of M. yunnanensis collected in Apr. 2009 in Honghe of Yunnan Province, P. R. China, and identified by Dr. De-Shan Deng. Voucher specimen (Cai091106) was deposited in the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.
Extraction and Isolation. Dried and powdered leaves and twigs of M. yunnanensis (40 kg) were extracted three times with methanol (MeOH) at room temperature and the solvent evaporated in vacuo. The residue was dissolved in 0.3% aqueous hydrochloric acid, and the solution subsequently basified using ammonia water to pH 9–10. The basic solution was partitioned with EtOAc, producing an aqueous and EtOAc phase. The resulting EtOAc fraction (205 g) was collected and then subjected to column chromatography over silica gel and eluted with a chloroform-acetone gradient (1/0 to 3/1, v/v) to afford five fractions (Ⅰ–Ⅶ), which were concentrated in vacuo. Fraction Ⅱ (5 g) was further chromatographed using a petroleum ether-acetone gradient (19/1 to 9/1, v/v) as the eluent to yield 8 subfractions, Ⅱ-1–Ⅱ-8. Compound 4 (46 mg) was crystallized from Ⅱ-8 (90 mg). Subfraction Ⅱ-1 (112 mg) was separated by semi-preparative reversed-phase C18-HPLC on a Sunfire column (19 × 250 mm) with a gradient flow of 70–75% aqueous MeOH to yield 2 (5 mg). Ⅱ-3 (120 mg) was further purified on a same semi-preparative column with a gradient flow of 65–80% aqueous MeOH to afford 5 (107 mg). Similar semi-preparative column separations with gradient flows of 60–75% aqueous MeOH were used to fractionate Ⅱ-4 (90 mg) and Ⅱ-6 (100 mg) to produce 1 (4 mg) and 3 (5 mg), respectively.
Meloyunine A (1): white powder; [α]D20 + 223 (c 0.16, MeOH); UV (MeOH) λmax (log ε) 247 (3.37), 278 (3.21) nm; IR (KBr) νmax 3431, 1702, 1610 cm-1; 1H and 13C NMR data, Table 1; ESIMS m/z 293 [M + H]+; positive ion HRESIMS m/z 293.1650 (calcd for C19H21N2O [M + H]+, 293.1653).
1H and 13C NMR data for meloyunines A-C (1-3) and 14, 15-didehydromelohenine B (4). (J in Hz, δ in ppm).
Meloyunine B (2): white powder; [α]D20 + 181 (c 0.15, MeOH); UV (MeOH) λmax (log ε) 246 (3.37), 280 (2.80) nm; IR (KBr) νmax 3433, 1703, 1613 cm-1; 1H and 13C NMR data, Table 1; ESIMS m/z 325 [M + H]+; positive ion HRESIMS m/z 325.1913 (calcd for C20H25N2O2 [M + H]+, 325.1916).
Meloyunine C (3): white powder; [α]D20 + 141 (c 0.13, MeOH); UV (MeOH) (log ε) λmax 230 (3.46), 292 (3.15) nm; IR (KBr) νmax 3431, 1708, 1623, 1592 cm-1; 1H and 13C NMR data, Table 1; ESIMS m/z 323 [M + H]+; positive ion HRESIMS m/z 323.1752 (calcd for C20H23N2O2 [M + H]+, 323.1759).
14, 15-Didehydromelohenine B (4): white powder; [α]D20 + 133 (c 0.06, MeOH); UV (MeOH) (log ε) λmax 228 (3.35), 298 (3.14) nm; IR (KBr) νmax 3483, 1686, 1668, 1598 cm−1; 1H and 13C NMR data, Table 1; ESIMS m/z 327 [M + H]+; positive ion HRESIMS m/z 327.1708 (calcd for C19H23N2O3 [M + H]+, 327. 1708).
Cytotoxicity Assay. Five human cancer cell lines, MCF-7 breast, SMMC-7721 hepatocellular carcinoma, HL-60 myeloid leukemia, SW480 colon cancer, and A-549 lung cancer, were used in the cytotoxic assay. Cells were cultured in RPMI-1640 or in DMEM medium (Hyclone, USA), supplemented with 10% fetal bovine serum (Hyclone, USA) in 5% CO2 at 37 ℃. The cytotoxicity assay was performed according to the MTT (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide) method in 96-well microplates.11 Briefly, 100 μL of adherent cells was seeded into each well of 96-well cell culture plates and allowed to adhere for 12 h before addition of test compounds, while suspended cells were seeded just before drug addition with initial density of 1 × 105 cells/mL. Each tumor cell line was exposed to the test compound at concentrations of 0.0625, 0.32, 1.6, 8, and 40 μM in triplicates for 48 h, with cisplatin (Sigma, USA) as positive control. After compound treatment, cell viability was detected and a cell growth curve was graphed. IC50 values were calculated by Reed and Muench's method.12
Notes
Electronic Supplementary Material
Supplementary material is available in the online version of this article at http://dx.doi.org/10.1007/s13659-011-0001-0 and is accessible for authorized users.
Acknowledgments
This work was supported in part by the National Natural Science Foundation of China (2107298), the 973 Program of Ministry of Science and Technology of P. R. China (2009CB522300), and Chinese Academy of Sciences (KSCX2-EW-R-15 and XiBuZhiGuang Project).
References
-
1.(a) Hutchinson, C. R. Tetrhedron 1981, 37, 1047-1065. (b) Sheriha, G. M. ; Rapoport, H. Phytochemistry 1976, 15, 505-508. PubMed Google Scholar
-
2.(a) O'Connor, S. E. ; Maresh, J. J. Nat. Prod. Rep. 2006, 23, 532-547. (b) Leete, E. ; Wemple, J. N. J. Am. Chem. Soc. 1969, 91, 2698-2702. (c) Battersby, A. R. ; Parry, R. J. J. Chem. Soc., Chem. Commun. 1971, 901-902. (d) Battersby, A. R. ; Brown, R. T. ; Kapil, R. S. ; Plunkett, A. O., Taylor, J. B. Chem. Commun. 1966, 46-47. PubMed Google Scholar
-
3.(a) Lorence, A. ; Nessler, C. L. Phytochemistry 2004, 65, 2735-2749. (b) Li, Q. Y. ; Zu, Y. G. ; Shi, R. Z. ; Yao, L. P. Curr. Med. Chem. 2006, 13, 2021-2039. (c) Thomas, C. J. ; Rahier, N. I. ; Hecht, S. M. Bioorg. Med. Chem. 2004, 12, 1585-1604. (d) Yamazaki, Y. ; Kitajima, M. ; Arita, M. ; Takayama, H. ; Sudo, H. ; Yamazaki, M. ; Aimi, N. ; Saito, K. Plant Physiol. 2004, 134, 161-170. PubMed Google Scholar
-
4.T. Feng, X. H. Cai, Y. Li, Y. Y. Wang, Y. Li, Y. P. Liu, M. J. Xie, X. D. Luo, Org. Lett. 11, 4834-4837 (2009) CrossRef PubMed Google Scholar
-
5.(a) Bernauer, K. ; Englert, G. ; Vetter, W. ; Weiss, E. Helv. Chim. Acta 1969, 52, 1886-1905. (b) Mehri, M. H. ; Rabaron, A. ; Sevenet, T. ; Plat, M. M. Phytochemistry 1978, 17, 1451-1452. (c) Rabaron, A. ; Mehri, M. H. ; Sevenet, T. ; Plat, M. M. Phytochemistry 1978, 17, 145-14532. (d) Baassou, S. ; Mehri, H. M. ; Rabaron, A. ; Plat, M. Tetrahedron Lett. 1983, 24, 761-762. (e) Mehri, M. ; Baassou, S. ; Plat, M. J. Nat. Prod. 1991, 54, 372-379. (f) Mehri, H. ; Plat, M. J. Nat. Prod. 1992, 55, 241-244. (g) He, Y. L. ; Chen, W. M. ; Feng, X. Z. J. Nat. Prod. 1994, 57, 411-414. (h) Zhang, Y. W. ; Yang, R. ; Cheng, Q. ; Ofuji, K. Helv. Chim. Acta 2003, 86, 415-419. PubMed Google Scholar
-
6.(a) Cai, X. H. ; Zeng, C. X. ; Feng, T. ; Li, Y. ; Luo, X. D. Helv. Chim. Acta 2010, 93, 2037-2044. (b) Cai, X. H. ; Shang, J. H; Feng, T. ; Luo, X. D. Z. Naturforsch., B 2010, 65, 1164-1168. (c) Cai, X. H. ; Feng, T; Liu, Y. P. ; Du, Z. Z. ; Luo, X. D. Org. Lett. 2008, 10, 577-580. (d) Cai, X. H. ; Du, Z. Z. ; Luo, X. D. Org. Lett. 2007, 9, 1817-1820. (e) Feng, T. ; Li, Y. ; Cai, X. H. ; Luo, X. D. J. Nat. Prod. 2009, 72, 1836-1841. (f) Feng, T. ; Cai, X. H. ; Zhao, P. J. ; Du Z. Z. ; Li, W. Q. ; Luo, X. D. Planta Med. 2009, 75, 1537-1541. PubMed Google Scholar
-
7.(a) Feng, T. ; Cai, X. H. ; Liu, Y. P. ; Li, Y. ; Wang, Y. Y. ; Luo, X. D. J. Nat. Prod. 2010, 73, 22-26. (b) Feng, T. ; Liu, Y. P. ; Cai, X. H. ; Wang, Y. Y. ; Luo, X. D. Org. Lett. 2010, 12, 968-971. (c) Feng, T. ; Li, Y. ; Wang, Y. Y. ; Cai, X. H. ; Liu, Y. P. ; Luo, X. D. J. Nat. Prod. 2010, 73, 1075-1079. PubMed Google Scholar
-
8.S Takano, S Hatakeyama, K Ogasawara, Heterocycles 6, 1311-1317 (1977) CrossRef PubMed Google Scholar
-
9.S. Baassou, H. Mehri, M. Plat, Ann. Pharm. Fr. 45, 49-56 (1987) PubMed Google Scholar
-
10.(a) Lanter, J. C. ; Sui, Z. H. ; Macielag, M. J. ; Fiordeliso, J. J. ; Jiang, W. Q. ; Qiu, Y. H. ; Bhattacharjee, S. ; Kraft, P. ; John, T. M. ; Haynes-Johnson, D. ; Craig, E. ; Clancy, J. J. Med. Chem. 2004, 47, 656-662. (b) Zhang, X. ; Jiang, W. ; Sui, Z. J. Org. Chem. 2003, 68, 4523-4526. PubMed Google Scholar
-
11.T. Mosmann, J. Immunol. Methods 65, 55-63 (1983) CrossRef PubMed Google Scholar
-
12.L. J. Reed, H. Muench, Am. J. Hyg. 27, 493-497 (1938) PubMed Google Scholar
Copyright information
© The Author(s) 2011
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
