 2022, Vol. 57
2022, Vol. 57

治疗2型糖尿病的药物可针对不同的环节和靶标, 其中包括胰高血糖素样肽-1 (glucagon-like-peptide-1, GLP-1) 受体激动剂。天然配体GLP-1 (7-37) (1) 是内源性肽, 具有控制血糖、改善β细胞功能、降低体重和心收缩压等功能。然而GLP-1在体内容易被代谢, 在血液中迅速被二肽基肽酶(DPP-4) 水解失活, 水解的特异性位点是Ala7-Glu8肽键。
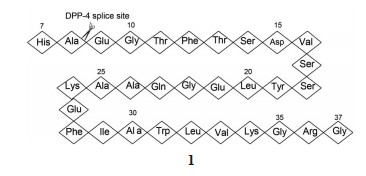
|
诺和诺德研发GLP-1受体激动剂, 已有一些同类药物上市, 表 1列出了药物名称和结构特征以及用药特点。艾塞那肽(2, exenatide) 是礼来上市的第一个GLP-1受体激动剂, 为毒蜥外泌肽去掉前8个氨基酸而成的活性形式His9-Ser47-NH2, 皮下注射, 半衰期为2.4 h, 虽然长于GLP-1, 但每日需给药两次。
| Table 1 The composition and dosage of marketed GLP-1 receptor agonists |
利拉鲁肽(3, liraglutide) 是诺和诺德2009年研发的肽类药物, 半衰期t1/2 11~15 h, 患者用特制注射器可自行每日皮下注射一次, 控制血糖于正常水平。其结构特征是将GLP-1的Lys34变换为Arg34, 并在Lys26共价连接出一个γ-Glu, 经棕榈酸酰化而成。长链的亲脂性有利于同白蛋白发生疏水性结合, 提高了药物的稳定性。
GSK研发的阿必鲁肽(4, albiglutide) 和礼来研发的杜拉鲁肽(5, dulaglutide) 是用基因工程方法分别与人白蛋白或IgG4重链Fc的融合蛋白, 显著提高了产品的稳定性, 半衰期7~8天, 达到每周给药一次。4和5是生物药。
诺和诺德在研发利拉鲁肽中, 对GLP-1各氨基酸残基的作用和亲脂长链的特征积累了丰富经验, 拟研发长效GLP-1受体激动剂, 途径是通过变化氨基酸残基和亲脂链的组成, 达到提高药效和稳定性、每周给药一次的目标。

|
用表达人GLP-1受体基因的幼仓鼠细胞(BHK cells) 膜为评价模型, 以不同浓度的受试物置换125I-GLP-1配体的能力, 计算化合物与受体的结合强度。由于不同亲脂链与白蛋白的结合能力不同, 对GLP-1的保护作用差异, 每个受试物的活性是在有2%白蛋白和没有白蛋白的条件下测定IC50, 以表征亲脂链对目标物稳定性的影响。
2.2 体外功能实验用表达人GLP-1受体和CRE荧光酶基因的幼仓鼠细胞模型, 评价化合物对离体细胞的功能性活性, 与不同浓度受试物温孵, 经处理后用TopCount NXT仪测定荧光读数, 经非线性回归计算化合物的EC50。
3 结构设计 3.1 亲脂性链长对结合与功能的影响研发长效激动剂目标是改善药代动力学性质, 但不降低活性强度。基于研发利拉鲁肽获得的构效关系和经验性规律, 将Lys34变换为Arg34, 并在Lys26的ζ氨基经Glu酰化, 后者连接出亲脂性脂肪酸链, 合成的代表性化合物列于表 2。
| Table 2 Structure, in vitro binding affinity and potency of typical compounds. HSA: Human serum albumin |

表 2的构效关系表明, 化合物6是GLP-1 (7-37) (1) 的Lys34被Arg34替换, 对受体的结合性能没有影响, 但激动功能提高1倍, 因而以后的优化都制备Arg34多肽。利拉鲁肽(3) 是在化合物6的Lys26经γ-Glu与棕榈酰基(C16酰基) 连接的分子, 白蛋白显著影响了6与受体的结合, 在有或没有白蛋白的存在下IC50比值为43, 系因亲脂性长链提高了利拉鲁肽与白蛋白的结合, 6的游离态分子减少的缘故, 但其功能与3相当, 是提高了稳定性的缘故。然而C18酰基(7) 的功能减弱, 不过长链的C20酰基若偶联亲水性的两个乙二醇(2OEG) 片段(8), 功能活性又显著提高, 提示亲脂链的长度和结构特征都对功能有影响。
3.2 Aib8、Arg34-GLP-1 (7-37) 脂肪链的变换鉴于GLP-1 (7-37) 被DPP-4水解的位点是His7-Ala8肽键, 为避免被水解, 将化合物6的Ala8变换成非天然的氨基异丁酸(Aib8, 12), 其结合性和功能保持不变。从而以12为母核[Aib8, Arg34-GLP-1 (7-37)], 连接不同的脂质链, 考察侧链对活性的影响, 如表 3所示。
| Table 3 SAR of derivatives of Aib8, Arg34-GLP-1(7-37) with C16-C20 acids and diacids attached to different linkers |

分析构效关系如下: ①化合物6的Ala8被氨基异丁酸替换为Aib8成12, 对受体结合作用未变, 激动功能略有提升。连接亲脂链的化合物13, 功能提高2倍多, 但仍略逊于利拉鲁肽(3)。②亲脂片段与激动剂之间的连接基(L) 对结合力和功能有显著影响。例如化合物17与只用γ-Glu连接的13相比, 功能提高了4.5倍, 存在2%白蛋白对结合力的影响降低了23倍, 提示增加了稳定性。在白蛋白存在下, 随着连接基的增长, 化合物与受体的结合力为17 > 16 = 15 > 13。③增长亲脂链对受体的结合作用有显著提升(无论白蛋白是否存在), 例如化合物18强于16, 但功能活性变化不大, 如化合物16、18和19的功能活性相近, 都高于利拉鲁肽(3), 提示连接基的延长是有益的。
化合物20~23的亲脂链末端含有羧基(由diacid合成), 目的是引入负电荷提高与白蛋白的碱性基团结合, 但末端羧基对结合性与功能没有改善, 反而降低。化合物23的功能比16弱2倍。
3.3 二酸类和连接基的优化以Aib8、Arg34-GLP-1 (7-37) 为核心骨架, 在Lys26处经不同的连接基连接C18二酸的亲脂链, 合成的化合物列于表 4。功能实验表明, 不同的连接基对活性的影响差异很大, 例如较简单的连接基γ-Glu、γ-Glu-OEG或γ-Glu-2OEG的活性EC50为5~10 pmol·L-1 (25~28), 而连接基benzyl-β-Ala-2OEG (30) 的活性减弱到5 990 pmol·L-1。连接基也影响对受体的结合能力, 在没有白蛋白存在时IC50从0.1 nmol·L-1到9 nmol·L-1, 相差70倍, 而加入2%白蛋白的结合活性差距加大, 比例的波动范围由2倍(30) 到940倍(28), 提示不同的连接基对白蛋白的结合力是不同的, 提供了优化稳定性的方向。
| Table 4 Compounds of Aib8, Arg34-GLP-1 (7-37) with C18-C20 diacids attached to different linkers |

优化至此, 获得了数个良好活性的化合物, 其中尤以化合物28的活性和稳定性突出, 不仅对细胞的功能活性和与受体的结合能力强, 而且与白蛋白的结合作用也强(有或无白蛋白的结合作用相差近千倍, 预示28的稳定性高)。然而这并没有证明C18二酸连接于Lys26为最佳的位置, 为此, 在GLP-1 (7-37) 肽链的不同位置用Lys替换氨基酸残基, 经不同的连接基连接C18二酸, 合成的化合物列于表 5。
| Table 5 Compounds of GLP-1 (7-37) peptides attached with C18 diacids via γ-Glu and OEG containing linkers and to 16, 22, 25, 26, 27, 36, 37 and 38 positions of the peptides |
结果表明, 功能活性低于10 pmol·L-1的化合物有37~39、41和46, 而且与受体结合的活性低于0.5 nmol·L-1, 然而这些化合物与白蛋白的结合作用不强, 比值超过100的化合物只有3个, 提示变化亲脂链的结合位置没有显示优势。
4 体内药代动力学和药效学评价 4.1 亲脂链对大鼠血浆暴露量的影响研发利拉鲁肽已证实化合物体外与受体的结合能力, 当加入白蛋白降低的越多(比值高), 体内血浆半衰期越长(Madsen K, Knudsen LB, Agersoe H, et al. Structure-activity and protraction relationship of long-acting glucagon-like peptide-1 derivative: importance of fatty acid length, polarity, and bulkness. J Med Chem, 2007, 50: 6126-6132)。为了证明在这个系列中也有这种相关性, 评价了化合物20(C12 diacid, γ-Glu-2OEG)、21(C14 diacid, γ-Glu-2OEG)、23(C16 diacid, γ-Glu-2OEG)、28(C18 diacid, γ-Glu-2OEG) 和49 (C20 diacid, γ-Glu-2OEG) 的大鼠血浆半衰期。化合物静脉注射测定48 h的血药浓度, 结果表明, 随着碳链的增长, 曲线下面积增大, 尤其是28(C18) 和49(C20) 的血浆暴露量显著高于C12~C16, 不过化合物49的体外比值85, 显著低于28(940)。
4.2 Aib8对大鼠血浆暴露量的影响配体GLP-1 (7-37) 对DPP-4的不稳定性是由于Ala8-Glu9是酶剪切的位点, 为了考察肽中残基Aib8-Glu9对体内稳定性的影响, 大鼠静脉注射利拉鲁肽(3, Ala8-Glu9, γ-Glu, C16)、11 (Ala8-Glu9, C18 diacid, γ-Glu-2OEG) 和28(Aib8-Glu9, C18 diacid, γ-Glu-2OEG) 测定在48 h的血浆暴露量, 结果表明, 28显著大于3和11, 3和11的曲线下面积相同, 说明Aib8置换Ala8残基提高了肽对DPP-4酶的稳定性。
基于化合物28的体内外活性和代谢稳定性, 确定为候选物, 定名塞马鲁肽(semaglutide) (Lau J, Bloch P, Schäffer L, et al. Discovery of the once-weekly glucagon-like peptide‑1 (GLP-1) analogue semaglutide. J Med Chem, 2015, 58: 7370-7380)。
4.3 对微型猪的体内药代动力学评价为了评价28是否为长效作用的GLP-1受体激动剂, 与每日一次用药利拉鲁肽作比较, 用微型猪作动物模型, 给药途径是静脉和皮下注射, 表 6列出了这两种给药途径的药代动力学数据。
| Table 6 Pharmacokinetic evaluation in Gőttingen mini-pigs following administration of semaglutide (28, 2 nmol·kg-1 i.v. or 2 nmol·kg-1 s.c.) and liraglutide (3, 0.5 nmol·kg-1 i.v. or 1.0 nmol·kg-1 s.c.) |
结果提示, 静脉注射利拉鲁肽的分布容积(Vd) 0.067 L·kg-1 (67 mL·kg-1), 非常接近于微型猪的血液体积65 mL·kg-1, 并且提示利拉鲁肽药物浓度迅速在血液与外周组织达到平衡。利拉鲁肽的清除率(CL) 为0.003 8 L·h-1·kg-1 (0.063 mL·min-1·kg-1), 半衰期(t1/2) 12.4 h。而塞马鲁肽的Vd为102 L·kg-1 (0.102 mL·kg-1), 比利拉鲁肽高1.5倍, 说明游离的塞马鲁肽在血液中有较低的浓度, 是因为与白蛋白结合较多的缘故, 塞马鲁肽的清除率(CL) 为0.001 6 L·h-1·kg-1 (0.027 mL·min-1·kg-1), 比利拉鲁肽低2倍, 半衰期(t1/2) 长3倍。皮下注射的平均存留时间(MRT) 塞马鲁肽是利拉鲁肽的3倍, 生物利用度(F) 高达94%。这些数据预示塞马鲁肽的长效性显著强于利拉鲁肽。
4.4 对db/db小鼠的降血糖作用db/db小鼠是2型糖尿病动物模型, 具有高血糖、高胰岛素血症和肥胖的生理生化特征。对一些高活性的化合物评价降血糖作用, 表明塞马鲁肽是最强的一个, 其降糖水平和持续时间显著强于利拉鲁肽(数据从略)。
基于上述体内外药效和药代性质, 塞马鲁肽(28) 进入临床研究, 经三期试验, 证明是注射或口服治疗2型糖尿病的长效药物, 每周用药一次, 为治疗糖尿病和减肥药物, 于2017年FDA批准上市。
5 塞马鲁肽母核与GLP-1受体结合的晶体结构塞马鲁肽分子与GLP-1受体的胞外域的复合物晶体结构难以达到衍射分析的要求, 因而制备了未经酰化的肽母核与受体胞外域的单晶。X射线衍射分析(1.8 Å) 提示, 其结合方式与天然配体GLP-1 (7-37) OH的模式完全相同, 这是因为氨基酸序列高度一致的缘故(Underwood CR, Garibay P, Knudsen LB, et al. Crystal structure of glucagon-like peptide-1 in complex with the extracellular domain of the glucagon-like peptide-1 receptor. J Biol Chem, 2010, 285: 723-730)。Lys26虽与受体的Glu128结合, 但该结合位点对活性贡献不大。以前的实验结果表明将Lys26变换成其他氨基酸残基对受体结合的影响不大, 提示将其酰化并连接出亲脂链对于受体结合没有明显影响, 这也是用没有亲脂链的肽母核制备单晶的依据。图 1是塞马鲁胺母核与GLP-1受体的胞外域复合物晶体部分结构图, 结合特征如下: ① GLP-1 (7-37) 的Lys34没有参与同受体的结合, 以柔性构象方式存在; 而变换成塞马鲁肽的Arg34采取了朝向Glu27的构象, 是经过水分子介导相互结合, 形成稳固的构象。②塞马鲁肽的Gly35和Arg36分别与本身骨架上的Leu32和Trp31的羰基形成氢键(图 1a)。③ Arg36的侧链取向于受体的Glu68和塞马鲁肽的Trp31之间的裂隙处, 在疏水相互作用中形成一个顶盖(图 1b)。

|
图 1 Crystal structure of the semaglutide peptide backbone (gray) in complex with the GLP-1 receptor extracellular domain; a: The structure of the C-terminus of the semaglutide peptide backbone. Hydrogen bond interactions are illustrated as dotted lines; b: Arg36 closes the hydrophobic ligand-receptor interface by aligning with Trp31 and Glu68. A water molecule is coordinated by Glu27 and Arg34 |
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和疗效评价等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多重性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
本刊曾刊登了诺和诺德研发的降血糖药物利拉鲁肽的研发路径, 作为胰高血糖素样肽-1受体(GLP-1R) 激动剂, 是对天然配体变换个别氨基酸并连接亲脂性脂肪链的改构物, 将配体的2 min的半衰期延长到12 h, 提高了活性和代谢稳定性, 每日用药一次。研发者在深入解析和应用构效关系的基础上, 进一步优化肽链、引入乙二醇连接基和优化脂肪二酸链, 研发出后续药物塞马鲁肽, 成为每周用药一次的长效药物, 可注射用也可口服, 同时具有减肥效用, 跟随性的塞马鲁肽超越了首创药利拉鲁肽。
(编者按)