 2022, Vol. 57
2022, Vol. 57



2. 武汉工程大学化学与环境工程学院, 湖北 武汉 430205
2. School of Chemistry and Environmental Engineering, Wuhan Institute of Technology, Wuhan 430205, China
冠状病毒最早于1937年从鸡的感染组织中被分离出来, 直到1965年第一株人类冠状病毒才被分离出来。由于冠状病毒在电子显微镜下可观察到其外膜上有明显的棒状粒子突起, 使其形态看上去像皇冠, 因此被命名为“冠状病毒”[1]。冠状病毒属于Nidovirus目, 分为4个属: α、β、γ和δ[2]。人类冠状病毒是具有正链单链RNA基因组的包膜病毒。目前, 已报道了7种人类冠状病毒, 其中HCoV-229E、HCoV-OC43、HCoV-NL63和HCoV-HKU1仅引起类似普通感冒的轻微呼吸道症状[3-5]。相较于上述4种病毒, SARS-CoV、MERS-CoV和SARS-CoV-2传染性强[6], 致死率高, 对人类健康和公共卫生造成了严重威胁[7, 8]。
冠状病毒主蛋白酶(Mpro, 3CLpro) 是位于冠状病毒非结构蛋白nsp5上的半胱氨酸蛋白酶, 包含大约300个氨基酸[9], 是大多数RNA病毒共同具有的蛋白酶, 负责催化冠状病毒前体多蛋白水解为病毒功能蛋白。同时Mpro具有较高保守性, 人体内没有同源类似物[10], 因此成为广谱抗冠状病毒药物的重要靶点[11, 12]。研究发现, 冠状病毒的主蛋白酶具有很多相似的结构和结合位点[13]。饶子和领衔的合作团队[14]和柳红/杨海涛/许叶春/张磊柯合作团队[15]分别给出了SARS-CoV 3CLpro (图 1a) 和SARS-CoV-2 3CLpro (图 1b) 与拟肽类抑制剂的结合方式(图 1)。这些抑制剂分子设计均是基于弹头(warheads) 策略, 弹头多数会和冠状病毒Mpro的半胱氨酸的巯基发生共价结合[16, 17], 少部分抑制剂为非共价结合抑制剂[18, 19]。本文根据弹头进行分类, 对抑制剂的分子结构、活性及其设计思路进行了综述。

|
Figure 1 Binding patterns of 3CLpro and peptidomimetic inhibitors[14, 15]. a: SARS-CoV 3CLpro; b: SARS-CoV-2 3CLpro |
以Mpro为靶点的冠状病毒抑制剂中, 拟肽类分子的设计策略中都包含有弹头。常用的弹头包括Michael受体、醛、酮、氰基等。
1.1 以Michael受体为弹头的拟肽类抑制剂Michael受体拟肽类抑制剂含有作为弹头的Michael受体, 是一类重要的半胱氨酸蛋白酶抑制剂。一般来说, Michael受体抑制剂的设计策略是用合适的Michael受体基团取代不同底物的酰胺键来得到目标分子。图 2描述了Michael受体基团使半胱氨酸蛋白酶失活的机制[20]。半胱氨酸残基与抑制剂的Michael受体基团进行1, 4-加成, 随后α-碳负离子质子化, 导致了该酶的不可逆失活。

|
Figure 2 The inactivation mechanism of cysteine protease[20] |
1999年, Dragovich课题组[21]基于三肽基人类鼻病毒(HRV) 主蛋白酶(3Cpro) 抑制剂(图 3) 设计了靶向HRV 3Cpro拟肽类抑制剂, 并对其结构片段P1、P2、P3和P4进行了优化, 得到的目标分子中以rupintrivir (AG7088, 图 3) 活性最佳(EC90 ≈ 0.1 μmol·L-1), 且细胞毒性低(CC50 > 100 μmol·L-1)。HRV是微小RNA病毒家族的成员, 是引起普通感冒的一个最重要的原因, 其Mpro (3Cpro) 和冠状病毒Mpro都属于半胱氨酸蛋白酶, 这些抑制剂的末端Michael受体通过与酶的活性位点半胱氨酸残基形成共价加合物, 不可逆地抑制Mpro, 从而显示出抗病毒作用。该课题组研究发现, 当P1 (图 3) 位置为内酰胺环时该类抑制剂显示出强的3C蛋白酶抑制活性。由于冠状病毒Mpro (3CLpro) 也为半胱氨酸蛋白酶, 类比可知, AG7088中共轭酯的部分也可作为Michael受体与3CLpro半胱氨酸残基共价结合[11], 因此, 研究者以AG7088为先导化合物展开了一系列冠状病毒Mpro抑制剂的设计合成。

|
Figure 3 Molecular design of AG7088. HRV: Human rhinovirus |
2004年, Knobler等[22]报道了AG7088在细胞水平对SARS-CoV无抑制作用。2005年, Shie等[23]对AG7088的P1、P3和P4位置进行结构优化, 并以荧光肽为底物, 通过计算荧光值来计算目标化合物的生物活性, 发现大部分目标化合物(三肽α, β-不饱和脂肪酸类) 都有一定的SARS-CoV Mpro抑制作用(IC50 = 11~39 μmol·L-1), 其中将AG7088的P1部分(内酰胺环) 改成苯环的化合物1 (图 4), 对SARS病毒的抑制作用达到13 μmol·L-1。同时通过酶-抑制剂复合物的计算机建模设计合成了一系列二肽类抑制剂(IC50 = 1~10 μmol·L-1), 其中化合物2 (图 4) 的酶水平抑制活性(SARS-CoV Mpro, Ki = 0.52 μmol·L-1) 和细胞水平抑制活性(Vero E6, EC50 = 0.18 μmol·L-1) 都表现优异, 且无明显细胞毒性(CC50 > 200 μmol·L-1)。

|
Figure 4 Structures and biological activity of compounds 1-5 |
2005年, 饶子和和马大为联合课题组[24]通过对SARS-CoV Mpro和猪传染性胃肠炎病毒(TGEV) Mpro的复合物晶体结构分析, 发现SARS-CoV Mpro具有高度保守的结合口袋, 并针对结合口袋, 设计并合成了一系列目标化合物, 通过同源建模进行初步的活性评价, 用荧光标记的底物来测试目标化合物的酶活性并验证同源模型的可靠性。其中, 化合物3 (图 4) 活性表现较好(SARS-CoV Mpro, Ki = 9.0 nmol·L-1)[25]。2006年, Hsu/Wang/Peters合作团队[26]设计合成了一系列目标化合物[对P2、P3和P4片段(图 4) 进行不同取代基替换来获得目标化合物], 其中4的抗病毒活性较高(SARS-CoV Mpro, Ki = 0.058 μmol·L-1; 229E Cell, IC50 = 0.88 μmol·L-1)。2007年, Ghosh课题组[27]根据SARS-CoV 3CLpro与抑制剂的复合物X射线晶体结构设计合成了一系列目标化合物, 基于荧光共振能量转移(FRET) 的微板法来进行生物活性评价, 发现在P4部位(图 4) 引入Boc-Ser基团会增强对SARS-CoV的3CLpro抑制活性(5: IC50 = 10 μmol·L-1, SARS-CoV 3CLpro)。
1.2 以醛为弹头的拟肽类抑制剂研究表明醛肽类化合物是大多数半胱氨酸蛋白酶的有效抑制剂。利用醛基作为弹头设计冠状病毒抑制剂被广泛认为是一种有效的策略。Mpro活性位点具有催化二联体(Cys-145/His-41), 醛肽类抑制剂可以可逆地结合SARS-CoV Mpro的活性位点巯基, 醛类抑制剂被催化二联体的半胱氨酸145攻击, 形成两种可能的非对映异构体(图 5)[28]。

|
Figure 5 Possible pathway of catalytic dimer cysteine 145 attacking aldehyde inhibitors[28] |
2006年Rademann课题组[29]以CMK (图 6, TGEV Mpro抑制剂) 与半胱氨酸残基的结合方式为基础, 设计了一系列针对SARS-CoV Mpro的肽醛化合物库, 通过虚拟筛选得到一系列目标化合物, 并采用了基于HPLC的检测方法定量测定目标化合物的生物活性。其中化合物AcESTLQ-H活性最高(SARS-CoV Mpro, IC50 = 7.5 μmol·L-1), 同时发现活性高的化合物与Mpro半胱氨酸残基的结合方式与规范模式不同(和CMK与半胱氨酸残基的结合方式类似)。同年, Hsu/Wang/Peters合作团队课题组[26]发表的文章中除了上文提到的Michael受体拟肽类抑制剂外, 还以醛基作为新的结合弹头设计了一系列目标化合物。通过人冠状病毒HCoV-229E和MRC-5细胞筛选系统以及抗SARS冠状病毒复制活性实验进行生物活性评价, 其中化合物7 (TG-0205221) 活性表现优秀(SARS-CoV Mpro, Ki = 53 nmol·L-1), 具有显著的抗SARS-CoV (IC50 = 0.6 μmol·L-1) 和HCoV-229E (IC50 = 0.14 μmol·L-1) 细胞活性。7与SARS-CoV Mpro的复合物晶体结构分析显示, 7与SARS-CoV Mpro间存在丰富的相互作用(包括共价键、氢键和大量疏水相互作用)。将化合物7与4相比较, 不难发现醛基相对于酯基而言, 可以更好的避免酯酶的水解作用, 从而保持构型, 起到抑制作用。同时将亮氨酸部分替换为了大体积的环己基甘氨酸, 环己基与亮氨酸的异丙基相比具有更强的疏水作用, 因此与结合口袋的结合更紧密, 于是改善了抑制剂的细胞活性。

|
Figure 6 Structures of compounds 6-8 |
2011年, Hilgenfeld课题组[28]对五肽醛肽类抑制剂进行了活性测试, 发现其具有SARS-CoV 3CLpro微摩尔的抑制活性, 通过对五肽醛与Mpro结合的4种复合物晶体结构的分析, 发现SARS-CoV Mpro的S2疏水口袋可以容纳丝氨酸甚至天冬氨酸侧链。因此该课题组进行了P1和P2位置(图 6, 化合物8) 的结构优化, 发现P1位置没有内酰胺环的目标化合物也具有活性。这些结果表明, 醛基作为弹头的拟肽类抑制剂时, SARS-CoV Mpro的S1和S2位点对抑制剂的P1和P2位置官能团具有更好的容忍性。
2018年, Groutas/Chang联合课题组[30]基于GC376与MERS-CoV结合的晶体结构, 通过结构引导设计合成了一系列含有哌啶环的广谱冠状病毒Mpro抑制剂, 并在R3和R4位置(图 7骨架Ⅱ) 进行取代基的选择优化, 通过FRET方法对目标化合物进行活性评价, 结果显示, 化合物9A活性表现优秀(SARS-CoV Mpro, IC50 = 5.1 μmol·L-1; MERS-CoV Mpro, IC50 = 0.4 μmol·L-1), 细胞毒性低(CC50 > 100 μmol·L-1), 且具有广谱抗冠状病毒作用。同时研究该类抑制剂与Mpro复合物晶体结构, 发现由于哌啶环的存在, 抑制剂分子延伸到了Mpro亚结合位点, 从而获得最佳药理学活性和药代动力学(PK) 特性。该研究给Mpro有利的亚结合位点的探索提供了帮助。2020年, Liu/Xu/Tseng/Fierke合作团队[31]基于SARS-CoV抑制剂GC376 (具有广谱抗病毒作用) 设计合成了一系列含β-(S-2-oxopyrrolidin-3-yl)-alaninal (Opal) 的SARS-CoV-2 Mpro抑制剂, 该类抑制剂均显示强的SARS-CoV Mpro结合效力(SARS-CoV-2 Mpro, Ki ≤ 100 nmol·L-1), 其中化合物9 (MPI3) 与SARS-CoV-2 Mpro的结合效力最高(Ki = 8.3 nmol·L-1)。

|
Figure 7 Design strategy of 9 and 9A |
2020年, Liu/Yang/Xu/Zhang合作团队基于饶子和/马大为联合课题组报道的SARS-CoV Mpro晶体结构(PDB ID: 2H2Z)[25], 通过分析抑制剂与Mpro结合口袋的作用模式, 设计并合成了两个优秀的化合物10、11 (图 8), 探究了这两个化合物与SARS-CoV-2 3CLpro的结合模式。抑制剂的P1部分可以与S1口袋形成氢键, 提高抑制剂的活性; S1′半胱氨酸的巯基与抑制剂P1′的结合弹头形成共价结合; P2部分引入疏水基团来提高抑制剂的生物活性, 同时需要比较大的基团来占据此空间; P3部分的杂环可以形成氢键, 增加和靶点的结合力[15]。化合物10和11在生物活性、细胞毒性以及药代动力学方面都显示是一个很好的先导化合物, 目前化合物10 (DC402234) 已进入Ⅰ期临床试验。

|
Figure 8 Design strategy of compounds 10 and 11[15] |
2021年, 杨胜勇/雷剑/郑永唐合作团队[32]基于SARS-CoV-2 Mpro晶体结构, 以及已经报道的CoVs Mpro抑制剂的优势片段, 设计合成了一系列以双环脯氨酸为核心骨架的化合物, 用FRET法测定了目标化合物对SARS-CoV-2 Mpro的生物活性, 发现所有化合物均有SARS-CoV-2 Mpro体外抑制活性(IC50 = 7.6~748.5 nmol·L-1), 12~17为其中代表(图 9)。其中化合物13抑制效果最佳(SARS-CoV-2 Mpro, IC50 = 7.6 nmol·L-1), 且没有细胞毒性(CC50 > 500 μmol·L-1), 是一个优秀的先导化合物。研究发现, 该类抑制剂的P2位置用脂肪稠环结构可以提高抑制剂分子的刚性与疏水性, 增强抑制剂分子的靶标结合力。同时, P3位置用合适大小的疏水芳香基取代, 可以进一步提高抑制剂的生物活性以及代谢稳定性。

|
Figure 9 Structures of compounds 12-17 |
2022年, 柳红/J. Neyts/许叶春/张磊珂合作团队[33]报道了针对SARS-CoV-2和肠病毒的广谱抑制剂研究。该课题组基于现有肠病毒3Cpro抑制剂, 经过分子杂交, 设计了如图 10两个系列的目标化合物, 并对所有目标化合物进行生物活性评价。结果显示, R1为吲哚结构的化合物18抑制活性最高(3CLpro: IC50 = 0.034 μmol·L-1, SARS-CoV-2: EC50 = 0.29 μmol·L-1), 且无细胞毒性(CC50 > 800 μmol·L-1)。同时, 与AG7088相比, 化合物18还表现出良好的药代动力学性质和更有效的抗冠状病毒活性, 是进一步开发抗冠状病毒药物的良好先导化合物。

|
Figure 10 Molecular hybridization design strategy of compound 18[33] |
2021年, Groutas/Chang合作团队[34]为了探索CoV Mpro结合口袋新的化学空间, 提高抑制剂与Mpro的相互作用, 在醛类抑制剂的结构中加入了一个构象受限的环己烷部分(图 11), 同时在抑制剂中引入了氘原子。研究发现, 环己烷部分与Mpro的S4口袋有很好的结合作用, 且引入的氘原子改善了抑制剂的PK特性, 提高了抑制剂的膜通透性。该类抑制剂都具有SARS-CoV-2 Mpro抑制活性(IC50 = 0.10~1.90 μmol·L-1) 和MERS-CoV Mpro抑制活性(IC50 = 49~63 nmol·L-1), 其中化合物19对Vero E6细胞的SARS-CoV-2抑制活性较高(EC50 = 35 nmol·L-1), 且无明显细胞毒性(CC50 > 100 μmol·L-1), 是开发抗冠状病毒药物的良好先导化合物。同年, Chang/Groutas/Kim合作团队[35]为了研究Thorpe-Ingold效应、氘化和立体化学在SARS-CoV-2 3CLpro高效无毒抑制剂设计中的应用, 在GC376的结构上引入氘原子和不同数目的氟原子, 来调节抑制剂的药理活性、药代动力学以及口服生物利用度, 使用双质粒筛选SARS-CoV-2 3CLpro抑制剂, 结果显示化合物34活性最佳(IC50 = 0.13 ± 0.04 μmol·L-1, SARS-CoV-2 Mpro; EC50 = 1.03 ± 0.47 μmol·L-1, Vero E6), 且无明显细胞毒性(CC50 > 100 μmol·L-1), 是一个优秀的先导化合物。

|
Figure 11 Structures of compounds 19-34 |
2004年, Vederas课题组[36]设计并合成了一系列邻苯二甲酰肼的酮-谷氨酰胺类似物, 并测试了它们对SARS-CoV 3CLpro的抑制作用。研究发现将三肽(Ac-Val-Thr-Leu) 附着在这些谷氨酰胺弹头上, 可以产生活性优秀的抑制剂(对SARS-CoV的抑制活性IC50 = 0.60~70 μmol·L-1), 其中代表性化合物35~42 (图 12)。不难发现, P1位置为内酰胺环结构的抑制剂活性普遍高于该位置为酰胺环结构。

|
Figure 12 Structures and biological activity of compounds 35-42 |
2012年, Chang/Kim合作团队[37]设计合成了不同弹头的二肽类似物抑制剂(图 13)。3种化合物对应3个弹头, 即β-酮酰胺(GC375, 43)、醛(GC373, 44) 和二硫基加合物(GC376, 45)。3种化合物对被测病毒都有很高的抑制效果(图 13中列出了其SARS-CoV 3CLpro抑制活性)。2020年, Ma课题组[38]使用基于FRET的酶促方法测定了几种抑制剂的抗SARS-CoV-2 Mpro的效力, 发现GC-376具有强效SARS-CoV-2 Mpro抑制活性(IC50 = 0.03 μmol·L-1), 无明显细胞毒性(CC50 > 100 μmol·L-1)。

|
Figure 13 Structures and biological activity of compounds 43-45 |
2020年, 柳红和Hilgenfeld联合课题组[39]为了获得高效的广谱抗α冠状病毒、β冠状病毒和肠道病毒的药物, 通过对SARS-CoV Mpro晶体结构[4, 7, 40] (PDB: 1UJ1、2BX3、2BX4) 分析, 进行了拟肽类α-酮酰胺类化合物的分子设计、合成与Mpro (3Cpro和3CLpro) 活性评价。结果显示, P2位置引入不同取代基(化合物46~54, 图 14) 对3个病毒的抑制活性影响趋势是类似的, 进一步说明3种病毒Mpro的相似性。其中化合物48不仅对MERS-CoV具有纳摩尔水平的抑制活性(EC50 = 0.4 ± 0.3 nmol·L-1, Huh7细胞), 而且对SARS-CoV具有微摩尔水平的抑制活性(EC50 = 2.1 ± 1.2 μmol·L-1, Vero E6细胞), 表现出广谱的冠状病毒抑制活性, 且细胞毒性较低(CC50 = 43 ± 2 μmol·L-1)。

|
Figure 14 Molecular design and biological activity of α-ketoamides |
针对化合物48的结构, 2020年, Hilgenfeld课题组[41]将P2与P3之间的酰胺键改造为吡啶酮内酰胺结构(图 15), 以提高该化合物在血浆中的半衰期, 从而更好地将该结构的先导化合物发展成为SARS-CoV-2 Mpro的有效抑制剂。结构优化后得到的代表性化合物55表现出良好的药代动力学特征, 并且具有明显的肺亲和性, 适合通过吸入途径给药。

|
Figure 15 Structure of inhibitor 55 containing pyridone |
2013年, Thanigaimalai等[42-44]针对SARS-CoV 3CLpro设计合成了一系列以苯并噻唑基酮为弹头的目标化合物, 并且通过对P3位置的刚性取代基进行变换, 得到了56~59 (图 16) 等目标化合物。活性测试结果显示, 目标化合物抑制活性(Ki, IC50) 均在亚微摩尔至纳摩尔。对体内药代动力学(吸收、分布、代谢、排泄及代谢) 的实验研究和理论计算结果表明, 化合物56具有发展成为治疗COVID-19药物的潜力[16]。

|
Figure 16 Structures and biological activity of compounds 56-59 |
2005年, Shie等[45]受到niclosamide对SARS-CoV抑制作用的启发, 制备了以硝基苯氨甲酰基为弹头的一系列目标化合物, 其中化合物60 (图 17) 的活性表现优异, 通过荧光十四肽底物检测了其对SARS-CoV 3CLpro的抑制活性(IC50 = 0.06 μmol·L-1, SARS-CoV)[46], 说明了以硝基苯氨甲酰基为弹头的可行性, 为拟肽类抑制剂弹头的多样性提供了思路。

|
Figure 17 Molecular design and biological activity of compound 60 |
在酮基的α位碳原子引入取代基, 一方面可以增强酮羰基与Cys145残基的共价结合能力; 另一方面增加的取代基可与结合口袋的其他氨基酸形成氢键, 从而增强抑制剂与蛋白酶的结合作用力。
2006年, Cai课题组[47]基于二肽的半胱天冬酶抑制剂(如MX1013) 设计了一系列N, N-二甲基谷氨酰氟甲基酮(Fmk) 类目标化合物。通过测量目标化合物对SARS-CoV诱导的细胞死亡的保护作用[细胞病变效应(CPE)], 以衡量化合物对SARS-CoV复制的抑制作用。结果显示, 在P2位置的不同取代基中, 异丁基取代的化合物61a (图 18) 活性最强(Vero, SARS-CoV 6109株, EC50 = 2.5 μmol·L-1), 同时在细胞中的毒性较低(EC50 > 100 μmol·L-1)。

|
Figure 18 Molecular design and biological activity of compound 61a-61d |
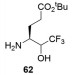
三氟甲基酮类化合物(TFMKs) 是丝氨酸[48]和半胱氨酸蛋白酶[49]抑制剂。2006年, Kiso/Hayashi合作团队[50]以化合物62 (表 1) 为原料通过5步合成了4个以TFMK为弹头的多肽类抑制剂(表 1, 63~66), 活性测试结果表明该类化合物的抑制效果不佳。2008年, Wong课题组[51]合成了一系列以TFMK为弹头的多肽类SARS-CoV 3CLpro抑制剂, 使用纯化的酶和荧光底物肽进行目标化合物的生物活性评估。结果显示(图 19), 67~69活性较高(IC50 = 10~20 μmol·L-1, SARS-CoV 3CLpro); 进一步的时间依赖性抑制研究表明, 69在4 h时抑制效果最佳(Ki = 0.3 μmol·L-1)。
| Table 1 Inhibitory activity of polypeptides against SARS-CoV 3CLpro |

|
Figure 19 Structures and biological activity of inhibitors 67-69 |
在拟肽类抑制剂的甲基酮弹头α位引入取代基, 卤素、羟基、乙酰氧基以及杂环的引入都有利于活性提高。2020年, Pfizer公司的Hoffman等[52]探究了拟肽类抑制剂酮甲基(弹头) 处的各种取代基对抑制剂抗病毒活性的影响, 最终确认了对SARS-CoV-2 3CLpro具有极强抑制活性的化合物70 (PF-00835231)。PF-00835231成药性较好, 是一种中度亲脂性(LogD7.4 = 1.7) 中性分子, 无可电离物质, 有望开发为用于静脉给药的COVID-19治疗药物。众所周知, 磷酸酯类前药可显著提高药物的水溶性[53, 54]。2021年, Pfizer公司的Boras等[55]又设计了化合物70的磷酸酯类前药71 (图 20), 磷酸盐前药呈弱酸性, LogD7.4 = -3.7 (预测值)。上述结果表明, 71的水溶性更佳, 临床医生在剂量方面具有更大的灵活性。

|
Figure 20 Structures of compounds 70-72 |
2022年, Nieman课题组[56]设计合成了以酰氧甲基酮为弹头的拟肽类SARS-CoV-2抑制剂, 通过CPE实验挑出针对不同冠状病毒的目标化合物, 再对这些目标化合物进行SARS-CoV-2抑制活性评价。其中72活性表现优秀(图 20, SARS-CoV-2 3CLpro, IC50 = 19 ± 0.5 nmol·L-1), 且无细胞毒性(CC50 > 200 μmol·L-1)。通过对酶-抑制剂复合物的晶体解析, 发现此类弹头的抑制剂也可和Cys145的残基共价结合, 进一步说明以取代的杂芳族和脂肪族α-酰氧甲基酮为弹头的拟肽类冠状病毒抑制剂的开发潜力。
1.7 以氰基为弹头的拟肽类抑制剂2013年, Wong课题组[57]设计并合成了4个以氰基为弹头的拟肽类抑制剂(73~76, 图 21), 并针对SARS-CoV Mpro通过使用荧光蛋白底物来测量目标化合物的生物活性。结果显示, 目标化合物的生物活性较强(IC50 = 4.6~49 μmol·L-1)。通过抑制剂与Mpro复合物晶体结构的分析, 发现抑制剂的氰基弹头也可以和催化位点Cys145残基产生共价作用, 从而起到抑制效果。

|
Figure 21 Structures of inhibitors 73-80 with a cyano warhead |
EV71 3Cpro抑制剂77 (图 21) 具有氰基弹头[58], 2019年进入Ⅲ期临床试验的组织蛋白酶K抑制剂odanacatib (78, 图 21) 也具有氰基弹头[59]。受此启发, Nieman课题组[60]于2021年设计了一系列以氰基为弹头的拟肽类SARS-CoV-2 3CLpro抑制剂。并在P3 (图 21, 化合物79) 位置进行了如图所示的结构优化, 同时对P1和P2位置了均尝试了两种取代基(P1: 五元内酰胺和六元内酰胺, P2: 异丙基和叔丁基), 用FRET法进行生物活性评价, 结果显示目标化合物的SARS-CoV-2 3CLpro抑制活性较高(IC50 = 9.6~49 nmol·L-1), 且大部分无明显细胞毒性(CC50 > 100 μmol·L-1), 化合物79a~79e (图 21) 为其中代表。研究发现氰基弹头对与人体组织蛋白酶S、B和L蛋白酶的选择性优于醛弹头。同年8月, 辉瑞公司在美国化学会2021年春季会议上公布了其口服COVID-19抗病毒临床候选药物nirmatrelvir (PF-07321332, 80), 该药物也是以氰基为弹头, 由此可知氰基弹头作为Mpro抑制剂的优越性[61]。2021年12月22日, 辉瑞公司宣布美国食品药品监督管理局(FDA) 已批准其新型COVID-19口服抗病毒候选药物Paxlovid (nirmatrelvir与ritonavir组成的复方制剂) 的紧急授权申请(EUA), 用于治疗非住院、具有发展成重症疾病高风险成人COVID-19感染。其中新冠病毒3CL蛋白酶抑制剂nirmatrelvir能通过阻断新冠病毒3CL蛋白酶的活性, 破坏新冠病毒的后续RNA复制过程; 另一种成分ritonavir则让nirmatrelvir在体内保持更长时间的活性, 更好地抗击病毒。令人鼓舞的是, 该药可以降低89%的死亡率, 不仅能够有效抑制德尔塔变异株复制, 对奥密克戎变异毒株也有效。
2 总结与展望当前, 中国的COVID-19疫情虽已得到有效遏制, 但由于新型冠状病毒在国外仍广泛流行, 外防输入的压力仍然较大, 国内也时有小规模疫情发生。当前国内外已有新冠疫苗上市, 大部分疫苗是针对SARS-CoV-2 S蛋白开发, 随着此病毒在全世界范围内的广泛传播, 除Alpha、Beta、Gamma、Delta和Omicron外, 新的变异毒株还在不断涌现, 而当前多种SARS-CoV-2变异株是由S蛋白突变导致, 因而使得疫苗作用有所下降[62]。最近一项针对新冠感染死亡患者的尸检报告揭示: 在所有44例样本中, 共涉及85个解剖位置和79种体液样本, 所有这些部位均有新冠病毒RNA检出, 直接证明新冠病毒不仅仅是破坏肺及呼吸道的正常组织结构, 还会扩散至全身各处[63]。
不论是2003年暴发的SARS-CoV、2012年暴发的MERS-CoV, 还是2019年底暴发的SARS-CoV-2, 都给人们生命健康、生产生活、经济发展和社会秩序带来了巨大挑战和严重影响; 在下一个10年是否会出现新的冠状病毒疫情仍充满不确定性, 只有开发出有效的广谱抗冠状病毒药物才能做到“心中不慌”。冠状病毒抑制剂先导化合物发现的药物化学策略包括老药新用、基于靶标的虚拟筛选、基于病毒蛋白结构的生物活性筛选、基于细胞水平的表型筛选、基于靶标的高通量晶体筛选、以靶标蛋白为微反应器的动态组合化学技术、基于侵入环节的肽模拟物设计和基于宿主的药物分子设计[64-69]。其中, 老药新用的优势是其安全性已经过验证, 可以绕过动物安全性研究(PK/PD和动物毒性实验), 直接进入临床Ⅱ或Ⅲ期研究, 并且能够保证供应。无疑, 在新疫情暴发时的应急药物研发, 药物筛选方法的建立是首要环节。然而在缺乏生物安全实验室和结构生物学等研究滞后的背景下, 计算机辅助药物设计作为现代药物研发的常规方法, 可加快特效药物的研究速度。针对特异性病毒靶标及宿主因子, 综合运用结构生物学、生物信息大数据、计算机辅助药物设计技术及药物设计新理念开展药物设计及化合物库筛选, 发现骨架新颖、结构多样的苗头化合物, 进而通过构效关系研究、结构优化及成药性评价, 发现先导化合物、候选药物及临床有效药物, 丰富抗病毒药物储备库, 以备抗“疫”之需[64]。
小分子口服药物在当前学术界和医药界被寄予厚望, 为终结疫情带来希望。靶向冠状病毒侵入过程、RNA复制过程、多聚蛋白裂解过程以及宿主靶标均可成为广谱冠状病毒抑制剂的靶标[70, 71]。默沙东公司开发的核苷类小分子口服药物molnupiravir是一种核苷类似物, 2021年11月4日英国批准molnupiravir上市, 这是全球首个获批用于治疗成人轻度至中度COVID-19的口服抗病毒药物。其作用靶酶RdRp结构保守, 使RdRp抑制剂在冠状病毒和埃博拉病毒等丝状病毒中具有相似的作用机制。因此, molnupiravir是一种潜在的广谱抗病毒药物。
冠状病毒的生命周期是一个复杂、连续、多步的过程, 病毒配体S蛋白、与病毒转录复制相关的RdRp、PLpro和3CLpro等多种蛋白质和酶发挥关键作用, 是治疗冠状病毒感染疾病的关键药物靶标[72-76]。在各种抗冠状病毒药物开发靶点中, Mpro是不同冠状病毒在复制和转录过程中的关键蛋白, 已成为抗冠状病毒药物研究中一个极为重要的研究靶点。靶向Mpro的拟肽类抑制剂在冠状病毒Mpro抑制剂表现尤为突出, 例如辉瑞公司的Paxlovid是FDA授权的首个COVID-19口服治疗药物, 其主要组分之一PF-07321332为拟肽类SARS-CoV-2抑制剂, 作用靶点为Mpro。开发可同时靶向冠状病毒其他靶标的多靶点药物(对因治疗和对症治疗), 开发拟肽类抑制剂和非拟肽类抑制剂的联合用药或分子杂合以发挥其协同作用, 从多个维度开发更安全、更有效和更佳药代动力学性质的抗冠状病毒药物, 也是开发冠状病毒抑制剂的有效策略。随着冠状病毒变种的不断出现, 抗冠状病毒药物研究必将吸引科学家和政府更多的研究投入, 相信靶向Mpro的小分子肽类抑制剂不仅会在抗击COVID-19疫情中发挥重要作用, 还会在广谱抗冠状病毒药物的开发中大放异彩。
作者贡献: 刘玉负责文献检索及综述撰写; 明巍负责图片修改和内容评估; 李陈宗负责综述中的数据核对和参考文献检查; 朱园园负责文章内容核对和审校; 古双喜负责为综述撰写思路并对稿件进行修改和审校。
利益冲突: 所有作者均声明不存在利益冲突。
| [1] |
Hamre D, Procknow JJ. A new virus isolated from the human respiratory tract[J]. Proc Soc Exp Biol Med, 1966, 121: 190-193. DOI:10.3181/00379727-121-30734 |
| [2] |
Pillaiyar T, Meenakshisundaram S, Manickam M. Recent discovery and development of inhibitors targeting coronaviruses[J]. Drug Discov Today, 2020, 25: 668-688. DOI:10.1016/j.drudis.2020.01.015 |
| [3] |
Tan GT, Kinghorn A, Hughes SH, et al. Psychotrine and its O-methyl ether are selective inhibitors of human immunodeficiency virus-1 reverse transcriptase[J]. J Biol Chem, 1991, 266: 23529-23536. DOI:10.1016/S0021-9258(18)54314-3 |
| [4] |
Anubhav K, Suresh S, Frank E. Human coronavirus-HKU1 infection among adults in Cleveland, Ohio[J]. Open Forum Infect Dis, 2017, 4: 1-6. |
| [5] |
Lau S, Woo P, Yip C, et al. Coronavirus HKU1 and other coronavirus infections in Hong Kong[J]. J Clin Microbiol, 2006, 44: 2063-2071. DOI:10.1128/JCM.02614-05 |
| [6] |
Zhu N, Zhang D, Wang W, at al. A novel coronavirus from patients with pneumonia in China, 2019[J]. N Engl J Med, 2020, 382: 727-733. DOI:10.1056/NEJMoa2001017 |
| [7] |
Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design[J]. FEBS J, 2015, 281: 4085-4096. |
| [8] |
Lu RJ, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding[J]. Lancet, 2020, 395: 565-574. DOI:10.1016/S0140-6736(20)30251-8 |
| [9] |
Usman B, Jennifer B, Adrian VC, et al. Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro[J]. Biochemistry, 2004, 43: 4906-4912. DOI:10.1021/bi0361766 |
| [10] |
Ma L, Zhao JY, Guo SS, 等. Establishment of a cell-based screening assay for inhibitors of SARS-CoV-2 3CL protease[J]. Acta Pharm Sin (药学学报), 2020, 55: 2122-2126. |
| [11] |
Zhao PP, Zheng WH, Bu M, 等. Progress in development of coronavirus inhibitors[J]. Chem Bull (化学通报), 2020, 83: 674-689. |
| [12] |
Yunjeong K, Liu H, Galasiti K, et al. Reversal of the progression of fatal coronavirus infection in cats by a broad-spectrum coronavirus protease inhibitor[J]. PLoS Pathog, 2016, 12: e1005531. DOI:10.1371/journal.ppat.1005531 |
| [13] |
Hu T, Liu Y, Zhao M, et al. A comparison of COVID-19, SARS and MERS[J]. PeerJ, 2020, 8: e9725. DOI:10.7717/peerj.9725 |
| [14] |
Yang H, Yang M, Ding Y, et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor[J]. Proc Natl Acad Sci U S A, 2003, 100: 13190-13195. DOI:10.1073/pnas.1835675100 |
| [15] |
Dai W, Zhang B, Jiang XM, et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease[J]. Science, 2020, 368: 1331-1335. DOI:10.1126/science.abb4489 |
| [16] |
Konno S, Kobayashi K, Senda M, et al. 3CL protease inhibitors with an electrophilic arylk-etone moiety as anti-SARS-CoV-2 agents[J]. J Med Chem, 2022, 65: 2926-2939. DOI:10.1021/acs.jmedchem.1c00665 |
| [17] |
Vankadara S, Yun XW, Liu B, et al. A head-to-head comparison of the inhibitory activities of 15 peptidomimetic SARS-CoV-2 3CLpro inhibitors[J]. Bioorg Med Chem Lett, 2021, 48: 128263. DOI:10.1016/j.bmcl.2021.128263 |
| [18] |
Kitamura N, Sacco MD, Ma C, et al. Expedited approach toward the rational design of noncovalent SARS-CoV-2 main protease inhibitors[J]. J Med Chem, 2022, 65: 2848-2865. DOI:10.1021/acs.jmedchem.1c00509 |
| [19] |
Han SH, Goins CM, Arya T, et al. Structure-based optimization of ML300-derived, noncovalent inhibitors targeting the severe acute respiratory syndrome coronavirus 3CL protease (SARS-CoV-2 3CLpro)[J]. J Med Chem, 2022, 65: 2880-2904. DOI:10.1021/acs.jmedchem.1c00598 |
| [20] |
Pillaiyar T, Manickam M, Namasivayam V, et al. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy[J]. J Med Chem, 2016, 59: 6595-6628. DOI:10.1021/acs.jmedchem.5b01461 |
| [21] |
Dragovich PS, Prins TJ, Zhou R, et al. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 4. Incorporation of P1 lactam moieties as L-glutamine replacements[J]. J Med Chem, 1999, 42: 1213-1224. DOI:10.1021/jm9805384 |
| [22] |
Knobler S, Mahmoud R, Lemon SM, et al. Learning From SARS: Preparing for the Next Disease Outbreak -- Workshop Summary[M]. Washington, D.C.: The National Academies Press, 2004.
|
| [23] |
Shie JJ, Fang JM, Kuo TH, et al. Inhibition of the severe acute respiratory syndrome 3CL protease by peptidomimetic α, β-unsa-turated esters[J]. Bioorg Med Chem, 2005, 13: 5240-5252. DOI:10.1016/j.bmc.2005.05.065 |
| [24] |
Anand K, Ziebuhr J, Wadhwani P, et al. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs[J]. Science, 2003, 300: 1763-1767. DOI:10.1126/science.1085658 |
| [25] |
Yang H, Xie W, Xue X, et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases[J]. PLoS Biol, 2005, 3: 1742-1752. |
| [26] |
Yang S, Chen SJ, Hsu MF, et al. Synthesis, crystal structure, structure activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor[J]. J Med Chem, 2006, 49: 4971-4980. DOI:10.1021/jm0603926 |
| [27] |
Ghosh AK, Kai X, Grum-Tokars V, et al. Structure-based design, synthesis, and biological evaluation of peptidomimetic SARS-CoV 3CLpro inhibitors[J]. Bioorg Med Chem Lett, 2007, 17: 5876-5880. DOI:10.1016/j.bmcl.2007.08.031 |
| [28] |
Zhu L, George S, Schmidt MF, et al. Peptide aldehyde inhibitors challenge the substrate specificity of the SARS-coronavirus main protease[J]. Antiviral Res, 2011, 92: 204-212. DOI:10.1016/j.antiviral.2011.08.001 |
| [29] |
Al-Gharabli SI, Shah S, Weik S, et al. An efficient method for the synthesis of peptide aldehyde libraries employed in the discovery of reversible SARS coronavirus main protease (SARS-CoV Mpro) inhibitors[J]. ChemBioChem, 2006, 7: 1048-1055. DOI:10.1002/cbic.200500533 |
| [30] |
Galasiti Kankanamalage AC, Kim Y, Damalanka VC, et al. Structure-guided design of potent and permeable inhibitors of MERS coronavirus 3CL protease that utilize a piperidine moiety as a novel design element[J]. Eur J Med Chem, 2018, 160: 334-336. |
| [31] |
Yang KS, Ma XR, Ma Y, et al. A quick route to multiple highly potent SARS-CoV-2 main protease inhibitors[J]. ChemMedChem, 2020, 15: 1-8. DOI:10.1002/cmdc.201900683 |
| [32] |
Qiao J, Li YS, Zeng R, et al. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model[J]. Science, 2021, 371: 1374-1378. |
| [33] |
Dai W, Jochmans D, Xie H, et al. Design, synthesis, and biological evaluation of peptidoimetic aldehydes as broad-spectrum inhibitors against enterovirus and SARS-CoV-2[J]. J Med Chem, 2022, 65: 2794-2808. DOI:10.1021/acs.jmedchem.0c02258 |
| [34] |
Chamandi SD, Yunjeong K, Naemi B, et al. Structure-guided design of conformationally constrained cyclohexane inhibitors of severe acute respiratory syndrome coronavirus-2 3CL protease[J]. J Med Chem, 2021, 64: 10047-10058. DOI:10.1021/acs.jmedchem.1c00319 |
| [35] |
Dampalla C, Rathnayake AD, Perera KD, et al. Structure-guided design of potent inhibitors of SARS-CoV-2 3CL protease: structural, biochemical, and cell-based studies[J]. J Med Chem, 2021, 64: 17846-17865. DOI:10.1021/acs.jmedchem.1c01037 |
| [36] |
Jain RP, Pettersson HI, Zhang J, et al. Synthesis and evaluation of keto-glutamine analogues as potent inhibitors of severe acute respiratory syndrome 3CLpro[J]. J Med Chem, 2004, 47: 6113-6116. DOI:10.1021/jm0494873 |
| [37] |
Kim Y, Lovell S, Tiew KC, et al. Broad-spectrum antivirals against 3C or 3C-Like proteases of picornaviruses, noroviruses, and coronaviruses[J]. J Virol, 2012, 86: 11754-11762. DOI:10.1128/JVI.01348-12 |
| [38] |
Ma C, Sacco M, Hurst B, et al. Boceprevir, GC-376, and calpain inhibitors Ⅱ, XⅡ inhibit SARS-CoV-2 viral replication by targeting the viral main protease[J]. Cell Res, 2020, 30: 678-692. DOI:10.1038/s41422-020-0356-z |
| [39] |
Zhang L, Lin D, Kusov Y, et al. Alpha-ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication[J]. J Med Chem, 2020, 63: 4562-4578. DOI:10.1021/acs.jmedchem.9b01828 |
| [40] |
Tan J, Verschueren K, Anand K, et al. pH-dependent conformational flexibility of the SARS-CoV main proteinase (Mpro) dimer: molecular dynamics simulations and multiple X-ray structure analyses[J]. J Mol Biol, 2005, 354: 25-40. DOI:10.1016/j.jmb.2005.09.012 |
| [41] |
Zhang L, Lin D, Sun X, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors[J]. Science, 2020, 368: 409-412. DOI:10.1126/science.abb3405 |
| [42] |
Thanigaimalai P, Konno S, Yamamoto T, et al. Design, synthesis, and biological evaluation of novel dipeptide-type SARS-CoV 3CL protease inhibitors: structure-activity relationship study[J]. Eur J Med Chem, 2013, 65: 436-447. DOI:10.1016/j.ejmech.2013.05.005 |
| [43] |
Thanigaimalai P, Konno S, Yamamoto T, et al. Development of potent dipeptide-type SARS-CoV 3CL protease inhibitors with novel P3 scaffolds: design, synthesis, biological evaluation, and docking studies[J]. Eur J Med Chem, 2013, 68: 372-384. DOI:10.1016/j.ejmech.2013.07.037 |
| [44] |
Konno S, Thanigaimalai P, Yamamotoa T, et al. Design and synthesis of new tripeptide-type SARS-CoV 3CL protease inhibitors containing an electrophilic arylketone moiety[J]. Bioorg Med Chem, 2013, 21: 412-424. DOI:10.1016/j.bmc.2012.11.017 |
| [45] |
Shie JJ, Fang JM, Kuo CJ, et al. Discovery of potent anilide inhibitors against the severe acute respiratory syndrome 3CL protease[J]. J Med Chem, 2005, 48: 4469-4473. DOI:10.1021/jm050184y |
| [46] |
Wu CJ, Jan JT, Chen CM, et al. Inhibition of severe acute respiratory syndrome coronavirus replication by niclosamide[J]. Antimicrob Agents Chemother, 2004, 48: 2693-2696. DOI:10.1128/AAC.48.7.2693-2696.2004 |
| [47] |
Zhang HZ, Zhang H, Kemnitzer W, et al. Design and synthesis of dipeptidyl glutaminyl fluoromethyl ketones as potent severe acute respiratory syndrome coronovirus (SARS-CoV) inhibitors[J]. J Med Chem, 2006, 49: 1198-1201. DOI:10.1021/jm0507678 |
| [48] |
Laplante SR, Bonneau PR, Aubry N, et al. Characterization of the human cytomegalovirus protease as an induced-fit serine protease and the implications to the design of mechanism-based inhibitors[J]. J Am Chem Soc, 1999, 121: 2974-2986. DOI:10.1021/ja983904h |
| [49] |
Peet NP, Burkhart JP, Angelastro MR, et al. Synthesis of peptidyl fluoromethyl ketones and peptidyl alpha-keto esters as inhibitors of porcine pancreatic elastase, human neutrophil elastase, and rat and human neutrophil cathepsin G[J]. J Med Chem, 1990, 33: 394-407. DOI:10.1021/jm00163a063 |
| [50] |
Sydnes MO, Hayashi Y, Sharma VK, et al. Synthesis of glutamic acid and glutamine peptides possessing a trifluoromethyl ketone group as SARS-CoV 3CL protease inhibitors[J]. Tetrahedron, 2006, 62: 8601-8609. DOI:10.1016/j.tet.2006.06.052 |
| [51] |
Shao YM, Yang WB, Kuo TH, et al. Design, synthesis, and evaluation of trifluoromethyl ketones as inhibitors of SARS-CoV 3CL protease[J]. Bioorg Med Chem, 2008, 16: 4652-4660. DOI:10.1016/j.bmc.2008.02.040 |
| [52] |
Hoffman RL, Kania RS, Brothers MA, et al. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19[J]. J Med Chem, 2020, 63: 12725-12747. DOI:10.1021/acs.jmedchem.0c01063 |
| [53] |
Yu J, Wang J, Zhang L, 等. Application of phosphates and phosphonates prodrugs in drug research and development[J]. Acta Pharm Sin (药学学报), 2013, 47: 5-18. |
| [54] |
Gu SX, Du JW, Ju XL, et al. A scalable one-pot process for the synthesis of florfenicol phosphodiester[J]. Org Process Res Dev, 2014, 18: 552-554. DOI:10.1021/op500038s |
| [55] |
Boras B, Jones RM, Anson BJ, et al. Discovery of a novel inhibitor of coronavirus 3CL protease for the potential treatment of COVID-19[J]. bioRxiv, 2021. DOI:10.1101/2020.09.12.293498 |
| [56] |
Bai B, Belovodskiy A, Hena M, et al. Peptidomimetic alpha-acyloxymethylketone warheads with six-membered lactam P1 glutamine mimic: SARS-CoV-2 3CL protease inhibition, coronavirus antiviral activity, and in vitro biological stability[J]. J Med Chem, 2022, 65: 2905-2925. DOI:10.1021/acs.jmedchem.1c00616 |
| [57] |
Chuck CP, Chen C, Ke Z, et al. Design, synthesis and crystallographic analysis of nitrile-based broad-spectrum peptidomimetic inhibitors for coronavirus 3C-like proteases[J]. Eur J Med Chem, 2013, 59: 1-6. DOI:10.1016/j.ejmech.2012.10.053 |
| [58] |
Mcclung MR, O'Donoghue ML, Papapoulos SE, et al. Odanacatib for the treatment of postmenopausal osteoporosis: results of the LOFT multicentre, randomised, double-blind, placebo-controlled trial and LOFT Extension study[J]. Lancet Diabetes Endocrinol, 2019, 7: 7899-7911. |
| [59] |
Wang Y, Cao L, Zhai Y, et al. Inhibition of enterovirus 71 replication by an α-hydroxy-nitrile derivative NK-1.9k[J]. Antiviral Res, 2017, 141: 191-100. |
| [60] |
Bai B, Arutyunova E, Khan MB, et al. Peptidomimetic nitrile warheads as SARS-CoV-2 3CL protease inhibitors[J]. RSC Med Chem, 2021, 12: 1722-1730. DOI:10.1039/D1MD00247C |
| [61] |
Halford B. Pfizer unveils its oral SARS-CoV-2 inhibitor[N]. Chem Eng News, 2021, 99: 7. https://cen.acs.org/acs-news/acs-meeting-news/Pfizer-unveils-oral-SARS-CoV/99/i13.
|
| [62] |
Mao YP, Bian DW. Bioinformatics analysis of mutations in the spike protein of SARS-CoV-2 and their effects[J]. Chin J Virol (病毒学报), 2020, 36: 1020-1027. |
| [63] |
Daniel C, Sydney S, Sabrina R, et al. SARS-CoV-2 infection and persistence throughout the human body and brain[J]. Nat Portfolio, 2022. DOI:10.21203/rs.3.rs-1139035/v1 |
| [64] |
Li J, Jiang XY, Xu SJ, 等. Medicinal chemistry strategies in seeking coronavirus inhibitors[J]. Acta Pharm Sin (药学学报), 2020, 55: 537-553. |
| [65] |
Gao S, Huang T, Song L, et al. Medicinal chemistry strategies towards the development of effective SARS-CoV-2 inhibitors[J]. Acta Pharm Sin B, 2021, 12: 581-599. |
| [66] |
Xu YC, Liu H, Li JF, 等. Recent progress in drug development against COVID-19[J]. Chin Biotechnol (中国生物工程杂志), 2021, 41: 111-118. |
| [67] |
Yan H, Sun J, Wang K, et al. Repurposing CFDA-approved drug carrimycin as an antiviral agent against human coronaviruses, including the currently pandemic SARS-CoV-2[J]. Acta Pharm Sin B, 2021, 11: 2850-2858. DOI:10.1016/j.apsb.2021.02.024 |
| [68] |
Huang L, Gu SX. Small molecular drugs for potential therapy of COVID-19 and their patent research[J]. Chin J Pharm (中国医药工业杂志), 2020, 51: 467-475. |
| [69] |
Liu QY, Wang XL. Strategies for the development of drugs targeting novel coronavirus 2019-nCoV[J]. Acta Pharm Sin (药学学报), 2020, 55: 181-188. |
| [70] |
Ji XK, Jiang XY, Li J, 等. Advances in protease-targeted antiviral drugs[J]. Chin J Med Chem (中国药物化学杂志), 2021, 31: 739-756. |
| [71] |
Song LT, Cheng YS, Gao SH, 等. Advances in research on broad-spectrum inhibitors of human coronaviruses[J]. Chin J Med Chem (中国药物化学杂志), 2021, 31: 721-738. |
| [72] |
Huang TG, Sun L, Zhan P, 等. Recent advances in the research of broad-spectrum antiviral agents[J]. Acta Pharm Sin (药学学报), 2020, 55: 157-171. |
| [73] |
Yao ZG, Xia CN. Research progress on small molecule inhibitors of coronaviruses[J]. J Pharm Pract (药学实践杂志), 2020, 38: 385-397. |
| [74] |
Yang L, Wang HQ, Li YH. Research progress on therapeutic drugs for corona virus disease 2019[J]. Acta Pharm Sin (药学学报), 2020, 55: 1081-1090. |
| [75] |
Wang XY, Peng C, Yang YQ, 等. Research progress on active compounds against pathogenic coronaviruses and their potential target proteins[J]. Acta Pharm Sin (药学学报), 2020, 55: 2340-2357. |
| [76] |
Liu AL, Du GH. Drug discovery for COVID-19 treatment based on drug targets[J]. Acta Pharm Sin (药学学报), 2020, 55: 1073-1080. |