 2022, Vol. 57
2022, Vol. 57

·新药发现与研究实例简析·
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多维性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
本文的篇幅较长, 是因为叙述了三个药物研发历程, 比卡鲁胺、恩杂鲁胺和阿帕鲁胺都是非甾类雄激素受体拮抗剂, 治疗前列腺癌, 它们的作用靶标和机制相同, 但后继的药物在一定程度上克服了前驱的不足, 例如不良反应和耐受性等, 因而贡献于不同的时段。三个“鲁胺”都是跟随性药物, 后来居上, 使原创的两个药物都已退出了市场。比卡鲁胺和恩杂鲁胺从不同的先导物出发, 结构变换和评价模式都不尽相同, 表现了首创色彩。有趣的是阿帕鲁胺与恩杂鲁胺的原研者是同一团队, 化学结构的较小变换, 治疗效果有显著的提高。 (编者按)
前列腺癌的发生和增殖与雄激素的刺激作用密切相关, 阻断雄激素作用是防治前列腺癌的生物学基础。手术去势可很大程度上切断雄激素来源, 但并不彻底, 因为肾脏也可合成雄激素。药物治疗的原则是抑制雄激素的生成, 或阻断雄激素受体功能的拮抗剂。甾体类抗雄激素治疗, 如醋酸环丙孕酮(cyproterone acetate) 有孕激素和促性腺激素作用等不良反应, 相似的甾体结构导致脱靶作用。促黄体激素释放激素的药物(LHRH类似物), 可抑制睾酮产生, 从而使血中睾酮浓度降到去势水平, 例如布舍瑞林、醋酸戈舍瑞林等。
本文叙述的3个雄激素受体拮抗剂为非甾体抗雄激素药物, 是研发抗前列腺癌药物的活跃领域, 虽然也有不良反应, 如男子乳房女性化, 以及阻止了雄激素对下丘脑的负反馈机制, 使得垂体提高了促黄体生成激素(LH) 的分泌, 后者刺激睾丸产生雄激素, 结果造成了拮抗剂诱导产生天然的激动剂的后果, 减弱了抗癌效果。当前研发雄激素受体拮抗剂的目标应是选择性地作用于性器官附件, 而不作用于中枢垂体LH。
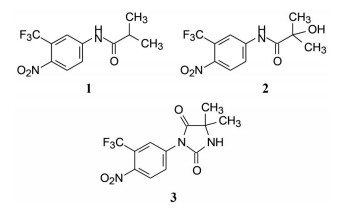
1.2 先驱的两个药第一个上市治疗前列腺癌的非甾体药物是氟他胺(1, flutamide), 作为雄激素拮抗剂, 它的发现带有偶然性。先灵药厂为研制抗菌药物而合成了硝基氟代苯胺类化合物, 却幸运地发现具有抗雄激素的作用(Baker JW, Bachman GL, Schumacher I, et al. Synthesis and bacteriostatic activity of some nitrotrifluoromethylanilides. J Med Chem, 1967, 10: 93-95)。随后经过临床前和临床研究, 证明具有抗雄激素和抑制前列腺癌的效果, 于1983年在德国上市, 6年后FDA也批准氟他胺在美国上市, 治疗前列腺癌。氟他胺是前药, 本身没有抗雄激素作用, 在体内被氧化代谢, 生成活性形式羟基氟他胺(2, hydroxyflutamie), 呈现对雄激素受体的拮抗作用。
Russel药厂研制的尼鲁米特(3, nilutamide) 是第二个非甾体类雄激素拮抗剂, 于1989年上市。化学结构与氟他胺有相同之处, 都含硝基氟代苯胺的片段, 不同的是以乙内酰脲片段连接于苯环上(Raynaud JP, Bonne C, Bouton MM, et al. Action of a non-steroid anti-androgen, RU 23908, in peripheral and central tissues. J Steroid Biochem, 1979, 11: 93-99)。氟他胺和尼鲁米特对雄激素受体的结合作用只有中等强度(Kd为亚微摩尔水平), 低于睾酮和二氢睾酮近百倍, 因而治疗剂量较大, 不良反应明显。
氟他胺和尼鲁米特是两个不同结构骨架的首创性药物, 在临床上的应用时间不长, 就被更好的跟随性药物所代替, 但两个药物在前列腺癌药物领域的地位不容忽视, 不仅在于从无到有的首创性, 而且后继上市药物都是以氟他胺或尼鲁米特为先导化合物而优化成功的。英国ICI公司研发的比卡鲁胺可视作跟随性药物, 迄今仍在临床应用。
|
研制比卡鲁胺和评价化合物的活性都是用整体动物模型, 分3个活性指标。
2.1.1 抗雄激素活性受试化合物以不同的剂量灌胃雄性大鼠, 每组5只, 每日一次, 连续4天, 最后一次给药24 h后牺牲大鼠, 剥离精囊, 洗净称重。同时设阴性对照组(给赋形剂) 和阳性对照组(灌胃氟他胺5 mg·kg-1), 阳性与阴性对照组的平均值之差为100%, 不同剂量的受试物组与阴性对照重量之差作图, 计算半数有效剂量ED50, 数值越小, 受试物的活性越强。
2.1.2 评价雄激素活性24日龄雄性大鼠去势后, 每日灌胃受试物25 mg·kg-1, 连续7日, 最后一次给药24 h后牺牲大鼠, 剥离精囊和前列腺, 称重, 计算手术去势对照组的重量差(mg), 差值越大表示对雄激素的激动作用越强。
2.1.3 评价选择性以3个剂量25、5和1 mg·kg-1灌胃雄性大鼠, 每日一次, 连续14天, 最后一次给药的24 h后, 取血测定血浆中黄体激素和睾酮水平, 并剥离和称重精囊。与只给赋形剂的对照组相比, 黄体激素和睾酮若没有变化, 表示具有选择性抗雄激素作用。
2.2 结构变换和构效关系ICI公司以氟他胺的代谢活化产物(2) 为先导物, 即将羟基预构于分子中。初步的结构变换发现苯环上两个亲电性取代基有利于活性, 而且在侧链末端引入大的疏水基团也增强活性, 因而对这些部位进行基团的变换和组合。
2.2.1 首轮设计探索各个位置的优势基团合成的代表性化合物对雄激素的拮抗/激动作用进行了评价, 结果列于表 1。构效关系分析如下: ① A环的3-和4-位为适宜的拉电子基团有利于抗雄激素活性, 如化合物5、11、14、17和19活性高于有甲基的21和单取代的10。② 4-位的优选基团是氰基(σp = 0.66) 或硝基(σp = 0.78), 3-位的优选基团是三氟甲基(σm = 0.43) 或氯原子(σm = 0.37), 表明4-位基团拉电子效应强于3-位。表现在化合物5 (4-CN, 3-CF3) 和8 (4-CN, 3-Cl) 比9 (4-Cl, 3-Cl) 活性高, 11 (4-CN, 3-CF3) 和14 (4-NO2, 3-CF3) 优于18 (4-Cl, 3-Cl), 19 (4-NO2, 3-Cl) 强于其位置异构体20 (4-Cl, 3-NO2)。③ R3为三氟甲基的系列化合物, 4-NO2, 3-CF3的活性并非占优, 例如化合物4不如R3为甲基的14。④连接基X在硫醚、亚砜和砜基之间变换, 考察硫的不同氧化态对活性的影响, 表明砜基连接的化合物似乎有优势, 亚砜是不稳定基团, 且增加了手性因素, 连接基的进一步优化是在硫醚和砜基之间。⑤苯环B的对位F取代的化合物(27、28、33和34) 的拮抗活性强于无取代或其他原子或基团取代的化合物。⑥大多数化合物未呈现对雄激素的激动作用。但R3为甲基和三氟甲基的化合物进行成对比较, 三氟甲基系列的雄激素的激动作用比较明显, 例如R3=CF3化合物5、8和23都有激动作用, 而相应的R3=CH3化合物11、17和24没有, 因此R3不宜为三氟甲基。
| Table 1 Structure and androgen antagonism / agonism of the typical compounds. a: The number quoted is the weight of the ventral prostate (mg); b: Percentage inhibition at the dose stated; c: NA indicates no statistically significant increase in ventral prostate weight compared to controls |
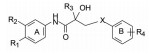
进而固定了R1和R2为最佳配置的4-CN, 3-CF3或4-NO2, 3-CF3, 以R3为三氟甲基, 变换侧链末端的取代基为各种烷基, 结果表明, 对雄激素的拮抗作用比较弱(数据省略)。
2.2.2 对R4的优化当优化出R1、R2和R3的优势结构后, 再考察R4片段, 为此, 将R1和R2固定为CN/CF3或NO2/CF3, R3为CH3, 变换R4为烷基或芳杂环。合成的化合物列于表 2。
| Table 2 Structure and androgen antagonism / agonism of the optimized R4 compounds |
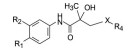
表 2的构效关系提示, 末端是烷基取代的化合物虽然没有刺激雄激素作用但拮抗活性较低。芳杂环如2-噻唑基(48) 和2-吡啶基(49) 的活性接近4-取代苯的活性, 推测适宜位置连接的极性芳杂环有利于提高拮抗活性。
2.2.3 选择性作用对于具有强抗雄激素活性且没有激动作用的化合物, 评价了选择性作用, 即化合物对于大鼠性器官附件的作用强于对下丘脑-垂体轴的作用, 以期对性器官的选择性抗性激素活性不会阻止天然雄激素对下丘脑-垂体轴的负反馈作用, 从而不诱导促黄体激素和睾酮的生成。代表性化合物的选择性作用列于表 3。
| Table 3 Effects on hormone levels on 14-day dosing in intact rats. Figures in the columns are the statistical significances for (a) the testosterone (TS) levels of the treated animals compared to a control group and (b) the luteinizing hormone (LH) levels of the treated animals compared to a control group; c: NS indicates no statistically significant difference from controls |
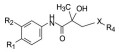
表 3是以3个剂量(25、5和1 mg·kg-1) 灌胃雄性大鼠14天, 与不给药的对照组比较, 化合物11、14、27、34、49和56对睾酮和促黄体激素水平没有影响; 结构的微小变化如49和50, 选择性明显变化。在这些化合物中, 化合物34活性强度、无激动雄激素性和不影响丘脑-垂体轴系统的选择性优胜于其他化合物。
2.3 候选化合物的确定和比卡鲁胺的上市进一步对化合物34的药效和药代与氟他胺作比较, 表 4列出的数据表明, 34活性与选择性显著强于氟他胺, 34的剂量19倍于ED50, 也不促进睾酮和促黄体激素的产生。
|
| Table 4 Comparison of PD and PK parameters of compound 34 and flutamide (1). a: Hormone levels following 14-day dosing to intact rats at 25 mg·kg-1 |
化合物34的代号为ICI 176334, 之后阿斯利康公司定名为比卡鲁胺(bicaluamide), 拆分后R和S都具有拮抗活性(R异构体代谢更慢), 故以消旋体进入临床前研究。临床研究表明, 比卡鲁胺口服应用, 与促黄体激素释放激素(GnRH) 类似物(如戈舍瑞林或亮丙瑞林) 合用或手术去势后用于治疗前列腺癌。美国FDA于1995年批准比卡鲁胺上市(Tucker H, Crook JW, Chesterson GJ. Nonsteroidal antiandrogens. Synthesis and structure-activity relationships of 3-substituted derivatives of 2-hydroxypropionanilides. J Med Chem, 1988, 31: 954-959)。
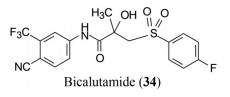
3 恩杂鲁胺的研制 3.1 概说恩杂鲁胺(59, enzalutamide) 是由美国Medivation公司开发, 2012年FDA批准上市。最初的研制是由UCLA大学的Jung和Memorial Sloan-Kattering癌症中心的Sawyers合作进行, 研发目标是对比卡鲁胺发生抵抗的前列腺癌的治疗药物。Jung等以药物尼鲁米特(3) 为先导物, 特别是以雄激素激动剂RU59063 (60) 为出发点, 改造结构成为拮抗剂。研发的路径和活性评价模式与比卡鲁胺不同, 从合成的200多个化合物中优选出恩杂鲁胺。
3.2 活性评价化合物的活性用3种体外方法评价。一是用对雄激素敏感的正常细胞LNCaP测定前列腺表面抗原(PSA) 表达读数; 另一模型是用携带有雄激素编码的cDNA病毒转染LNCaP细胞, 形成的LNCaP/AR细胞, 其雄激素受体表达水平提高3~5倍, 作为模拟去势后发生抵抗的前列腺癌细胞; 还用荧光素酶报告系统定量地测定大鼠前列腺中雄激素受体的活性。3种指标评价化合物对正常细胞、高表达雄激素的细胞和对雄激素受体转录功能的拮抗作用。
|
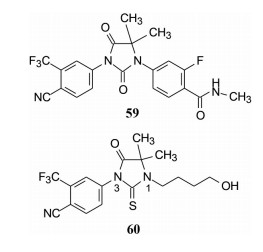
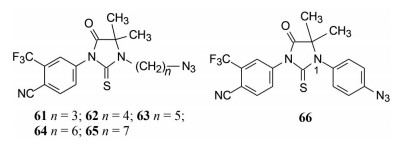
将化合物60的羟基用叠氮替换并以不同长度的碳链连接, 希望得到与雄激素受体结合更强的分子。合成的化合物61~66以DMSO (空白) 和比卡鲁胺(阳性) 为对照, 在正常的LNCaP细胞模型上测定PSA活性。结果表明这6个叠氮化合物活性都强于比卡鲁胺, 芳香叠氮物(66) 活性最强。从而开启了N1-取代芳环的硫代乙内酰脲的结构模式, 也因此与比卡鲁胺的结构类型不同。
|
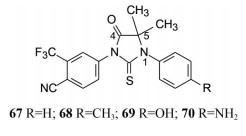
以66为新起点, 合成了N1-p-取代苯化合物67~70, 用PSA相对活性和荧光素酶报告系统评价活性, 表明p-甲基苯(68) 的活性最强。遂固定N1-甲苯基考察杂环的5, 5-二甲基的变换对活性的影响。
|
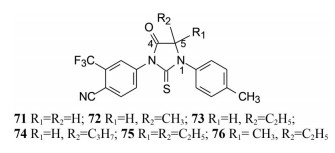
将N1固定为p-甲苯基, 变换硫代乙内酰脲5位的甲基, 合成单烷基和双烷基71~76, 评价荧光素酶活性的变化, 5位单烷基或尺寸大的双烷基活性都低于5, 5-二甲基化合物68。将5位双烷基与4位的氧代作位置互换, 活性仍低于68。
|
合成的5, 5-亚烷基化合物77~81 (螺丁烷~螺辛烷化合物), 以比卡鲁胺和68作对照, 评价抑制PSA的活性, 表明螺丁(77) 和螺戊(78) 的活性与68相当, 强于比卡鲁胺, 而大于五元螺环的活性较低。螺哌啶化合物82的活性尤差, 推测是哌啶N原子质子化极性加大不利于与受体结合。
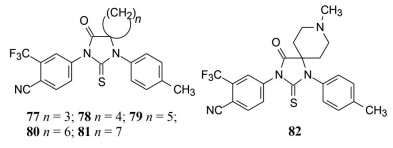
|
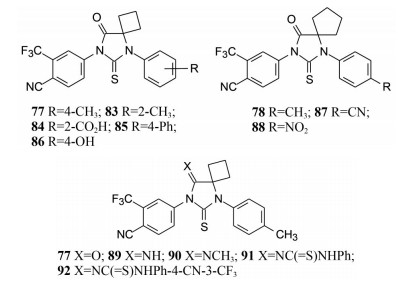
化合物77的4-甲基移至2-甲基(83), 抑制PSA的活性不变, 但2-羧基化合物84的活性显著降低, 提示带电荷的基团不利于结合。4-苯基(85, 为联苯) 和4-羟基(86) 的活性与77相同。螺戊环化合物的苯环对位氰基或硝基取代(87、88) 的活性与甲基(78) 相同, 提示只要5, 5-为螺丁或螺戊环, N1-苯环对位不同基团的变换, 仍保持活性。
3.3.6 乙内酰脲的酮基变换以优化的N1-甲苯基-5-螺环丁基为骨架, 变换4-酮基为(取代的) 亚胺, 合成的化合物89~92用PSA度数和荧光素酶评价活性(与化合物68和77比较), 结果表明亚胺类仍保持活性, 但91和92在细胞内不稳定, 水解生成77。
|
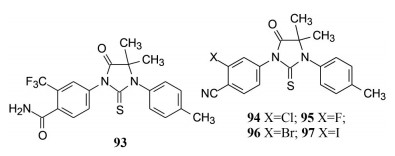
优化至此, 多种模型评价表明N1为p-甲基苯、5位是偕甲基或螺环丁基或螺环戊基为优选片段。下一步是固定这些优势基团, 探索变换N3的苯基取代基对活性的影响, 合成的化合物93 (氰基变为4-酰氨基) 和94~97 (三氟甲基变为卤素) 都失去了抗雄激素的活性, 进一步佐证了3-CF3-4-CN是优选基团, 这两个取代基不宜变换。
|
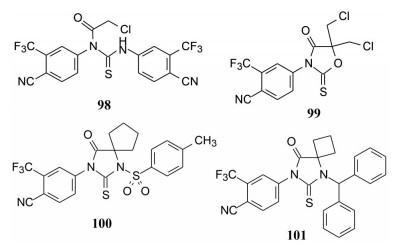
此外, 剖裂杂环成N1, N3-二苯基氯乙酰基硫脲化合物(98), 杂环变为噁唑硫代二酮(99), N1-苯基之间插入磺酰基(100) 以及N1-二苯甲基化合物(101) 都失去了活性, 佐证了前述优化路径得出的构效关系的结论。
|
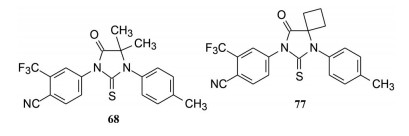
上述的优化过程, 68和77的体外活性突出, 尤其对模拟耐药的前列腺癌细胞LAPC4/AR的活性显著强于比卡鲁胺。用移植LAPC4/AR细胞的小鼠模型, 68和77的抑制效果显著强于比卡鲁胺。77抑制小鼠PSA分泌的IC50为124 nmol·L-1。然而77的药代动力学性质有缺陷, 在动物体内半衰期短, 静脉注射的t1/2小于60 min, 可能是由于高亲脂性(log P 4.20, 而比卡鲁胺log P 2.91) 苯环上甲基被代谢羟基化的缘故。因而进一步结构优化, 在保持活性的前提下降低分子的亲脂性和提高代谢稳定性, 目标聚焦于对位变换极性取代基。
|
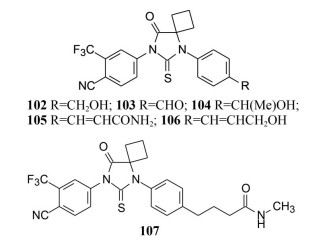
首先合成了具有极性基团取代的简单化合物102~104和两个插烯物105和106, 这些化合物的活性都很高, 例如羟甲基化合物102和104抑制PSA的IC50值分别为200和300 nmol·L-1, 苯甲醛化合物103的活性也很高, 笔者认为醛基为强亲电性基团, 有潜在的脱靶风险。两个插烯物105和106的活性更强, IC50值为100~150 nmol·L-1。遂后又合成了末端为羟基或酰胺基的长链分子, 活性仍很高(结构从略)。其中一个高活性化合物107的IC50值92 nmol·L-1。但脂溶性强(log P 3.44), 药动学性质较差, 分析原因是苯环上的烷基链易发生氧化代谢(如同化合物77)。
|
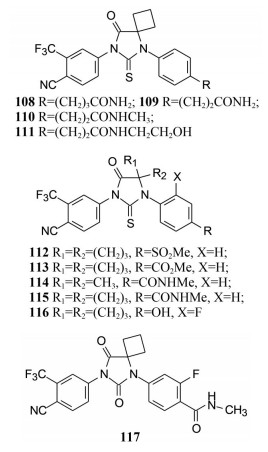
为此合成了化合物108~111, 碳链减少一个碳原子仍保持对激素抵抗的LAPC4/AR细胞产生PSA的抑制活性, 而化合物108和比卡鲁胺则没有活性。进而合成了化合物砜112、酯113、酰胺114和115, 以及氟代酚116, 都具有抑制LAPC4/AR的良好活性。
更为重要的是, 合成的化合物117, 不仅体外对LAPC4/AR细胞有强效的抑制PSA的活性(IC50 = 122 nmol·L-1), 而且有良好的药代动力学性质。表 5列出了代表性化合物的活性、clog P和灌胃动物同重量的受试物在稳态下的血药浓度。
|
| Table 5 The activity, clog P and plasma concentration of typical compounds. a: At steady state |
比较表 5化合物的活性显示, 117与77和107的IC50相近, 大约强于比卡鲁胺8倍, 物化性质如clog P适中。药动学上, 动物给受试物10 mg·kg-1, 化合物117稳态下的血药浓度与比卡鲁胺相同, 分别为9.9和10.0 mmol·L-1, 提示117的PD/PK优胜于比卡鲁胺, 更强于77和107 (Tran C, Ouk S, Clagg NJ, et al. Development of second-generation of antiandrogen for treatment of advanced prostate cancer. Science, 2009, 324: 797-790)。
3.5 确定候选物和恩杂鲁胺的上市化合物107是里程碑式分子, 继续优化的操作, 是固定N1-3-氟-4-甲酰甲胺基苯, N3为3-三氟甲基-4-氰基苯, 变换C5的烷基, 合成了化合物118和119, 以探索适配于新构效关系的C5取代。结果表明(图 1), 118和119抑制LAPC4/AR细胞分泌PSA的活性与117相同, 分析剂量-效应的关系, 化合物118的活性略优于117, 而且合成118比较容易, 起始原料丙酮比制备117的环丁酮方便易得。经系统药动学研究, 表明118的生物利用度F = 89.5%, 血浆蛋白结合率为97%, 半衰期t1/2 = 5.8日。再经安全性研究后, 确定118为候选化合物, 定名为恩杂鲁胺(enzalutamide), 转由Medication公司注册继续研发, 经Ⅲ期临床试验, 证明对比卡鲁胺治疗前列腺癌产生耐药的患者有效, 成为第二代治疗非转移性去势后抵抗性前列腺癌药物。于2012年经FDA批准上市(Jung ME, Ouk S, Yoo D, et al. Structure-activity relationship for thiohydantoin androgen receptor antagonists for castration-resistant prostate cancer (CRPC). J Med Chem, 2010, 53: 2779-2796)。
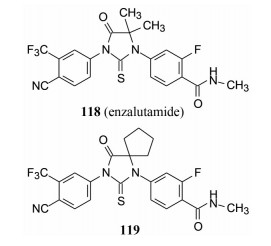
|

|
Figure 1 Comparison of the inhibitory activity of 117, 118 and 119. PSA: Prostate specific antigen |
比卡鲁胺和恩杂鲁胺作为去势后抵抗性前列腺癌(CRPC) 的治疗药物, 表现的主要缺点是剂量限制。比卡鲁胺在动物模型上对雄激素受体有部分激动作用, 限制了临床加大治疗剂量; 恩杂鲁胺可诱导雄激素受体在细胞核定位或与DNA结合的活性, 而且对中枢GABAA受体呈现一定的拮抗作用, 有引起癫痫的风险。因而, 恩杂鲁胺的发明者Jung和Sawyers继续研发更新的超越恩杂鲁胺的药物。
由于恩杂鲁胺分子的乙内酰脲杂环和N1的取代苯环都是优势片段, 只剩下对N3连接的苯环片段的变换。其专利披露了多种取代的芳杂环, 其实在实施例中只有两个化合物, 即120和121 [Jung ME, Sawyers CL, Ouk S, et al. Androgen receptor modulator for the treatment of prostate cancer and androgen receptor-associated diseases, WO2007126765 (A2)]。这两个化合物都与恩杂鲁胺的活性相近, 120是里程碑化合物77的吡啶等排体, 由于N1片段的优势显然120强于77, 化合物120是在2018年上市的阿帕鲁胺(apalutamide), 经转让给强生下属的杨森公司开发而获得成功。
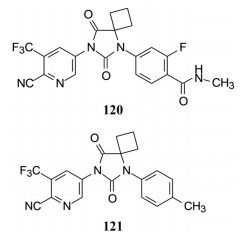
|
化合物120与117结构非常相似, 只是左侧的取代的苯环换成吡啶, 其余结构完全相同, 这是芳环等排置换的成功范例。表 6列出了120与比卡鲁胺和恩杂鲁胺的体外活性和一些药动学参数。可以看出3个药物的药代参数没有显著差别, 对雄激素受体的体外抑制活性120和118没有显著差异(二者强于比卡鲁胺8~10倍)。
| Table 6 Anti-androgen activity and PK values of typical compounds |
但体内实验对小鼠移植LAPC4/AR细胞的前列腺癌模型120显著优于118, 无论是单剂量实验, 或是多次给药血浆中达到稳态药物水平的实验, 120效果都强于118, 在较低稳态血药浓度和脑内浓度下120可产生最大治疗效果, 对患者预示有较大的治疗指数(宽治疗窗) 和降低癫痫等不良反应的风险。经临床前实验后, 阿帕鲁胺(120) 于2012年进入临床研究(Clegg NJ, Wongvipat J, Joseph JD, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res, 2012, 72: 1494-1503), Ⅲ期临床研究表明, 为口服治疗非转移性去势抵抗性前列腺癌的有效药物, 于2018年2月FDA批准在美国上市。