 2022, Vol. 57
2022, Vol. 57

间质表皮转化因子(c-mesenchymal-epithelial transition factor, c-Met) 是受体酪氨酸激酶的一种, 其配体是肝细胞生长因子(HGF), HGF与c-Met有高度的亲和力。HGF/c-Met信号通路调控多种生物功能, 例如细胞增殖、迁移、存活和凋亡以及创伤愈合和血管生成等, 较早就被发现在肿瘤细胞的活动中起到重要作用, c-Met激酶在多种肿瘤细胞中高表达, 而且常常发生突变, 例如晚期非小细胞肺癌(NSCLC) 患者发生c-Met基因外显子14跳跃突变。
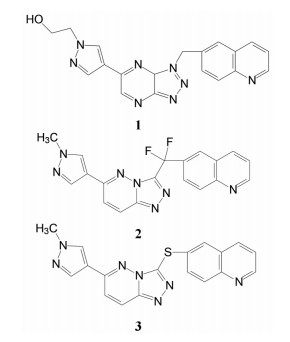
1.2 c-Met抑制剂的研究状态和黄医药启动项目研究时, 国际上已有针对c-Met激酶的ATP结合位点的研究, 第一阶段如葛兰素的候选化合物XL880 (Qian F, Engst S, Yamaguchi K, et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res, 2009, 69: 8009-8016) 和BMS的化合物BMS-777607 (Schroeder GM, An YM, Cai ZW, et al. Discovery of N-(4-(2-amino-3-chloropyridin-4-yloxy)-3-fluorophenyl)-4-ethoxy-1-(4-fluorophenyl)-2-oxo-1, 2-dihydropyridine-3-carboxamide (BMS-777607), a selective and orally efficacious inhibitor of the Met kinase super family. J Med Chem, 2009, 52: 1251-1254), 虽然都进行了深入的研究, 但由于是泛激酶抑制剂, 缺乏选择性而终止了研究。第二阶段的研究特征是提高了对c-Met的选择性, 有数个候选化合物进入了临床试验, 如辉瑞的PF04217903 (1) (Cui JJ, McTigue M, Nambu M, et al. Discovery of a novel class of exquisitely selective mesenchymal-epithelial transition factor (c-MET) protein kinase inhibitors and identification of the clinical candidate 2-(4-(1-(quinolin6-ylmethyl)-1H-[1,2,3]triazolo[4, 5-b]pyrazin-6-yl)-1H-pyrazol-1-yl)-ethanol (PF-04217903) for the treatment of cancer. J Med Chem, 2012, 55: 8091-8109)、强生的JNJ-38877605 (2) (Underiner TL, Herbertz T, Miknyoczki SJ. Discovery of small molecule c-Met inhibitors: evolution and profiles of clinical candidates. Anti-cancer Agents in Med Chem, 2010, 10: 7-27) 和SGX公司的SGX523 (3) (Buchanan SG, Hendle J, Lee PS, et al. SGX523 is an exquisite lyselective, ATP-competitive inhibitor of the MET receptor tyrosine kinase with antitumor activity in vivo. Mol Cancer Ther, 2009, 8: 3181-3190), 都对c-Met具有较高的选择性抑制作用, 从化学结构分析, 它们都具有喹啉环。然而在临床Ⅰ期试验中发现受试者的血浆中肌酐明显升高, 提示有肾脏毒性而终止研发。这三个临床失败的选择性抑制剂化学结构具有相似性: 中间的多氮杂环, 两侧的吡唑和喹啉环。
然而就在和黄医药研发本品即将完成之际, 2020年3月默克药厂的特泊替尼(4, tepotinib) 在日本上市, 是全球首款用于治疗不可切除的携带c-Met基因外显子14跳跃突变的晚期非小细胞肺癌(NSCLC) 患者的药物, 同年5月诺华的卡马替尼(5, capmatinib) 在美国上市, 也是治疗c-Met突变的晚期NSCLC药物。这是美国批准的针对这一患者群的首款靶向疗法。5的结构特征与前述的终止研发的相似, 但结构的变换消除了代谢产物引起的肾毒性。
|

|
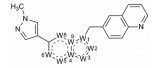
研制者启动项目时还没有针对c-Met为靶标的药物上市, 他们借鉴了安进公司研究的c-Met抑制剂6, 抑酶活性IC50 = 0.12 μmol·L-1 (Albrecht BK, Harmange JC, Bauer D, et al. Discovery and optimization of triazolopyridazines as potent and selective inhibitors of the c-Met kinase. J Med Chem, 2008, 51: 2879-2882), 公布的复合物晶体结构如图 1所示。结构显示6 (蓝色) 处于ATP结合腔中, 呈U形环绕着残基Met1211; 酚羟基与Met1160的NH发生氢键结合; 母核三唑并哒嗪环与Tyr1230芳环呈T型的π-π相互作用。化合物3~5的结合模式与此相似。

|
Figure 1 Co-crystal structure of 6 with c-Met |
用Transcreener荧光偏振ADP检测仪(Transcreener FP ADP Assay) 测定酶反应产生的ADP量, 方法是一定量的重组人c-Met加入缓冲液中, 加入受试物的系列稀释液, 然后加入ATP, 定时定温温孵后, 加入ATP检测液, 再定时定温温孵, 终止反应, 在荧光偏振仪的激光波长610 nm, 发射光波长670 nm下测定系列受试物影响c-Met产生的ADP的含量, 求出抑制活性IC50。
2.2.2 半体内测定对磷酸化c-Met (p-Met) 的抑制活性人非小细胞肺癌细胞NCI-H441定量地接种到雌性裸鼠, 两周后肿瘤体积为300 mm3, 随机分成两组, 一组灌胃受试物, 一组空白对照, 6 h后处死小鼠, 取出肿瘤组织, 经细胞裂解液处理, 细胞匀浆, 离心, 上清液用Bio-Rad测定总Met蛋白含量, ELISA测定p-Met含量。计算受试物对p-Met的抑制活性IC50。
2.2.3 体内活性和药代动力学测定用雌性裸鼠移植U87MG瘤细胞按常规方法测定受试物的抑瘤率(方法从略)。化合物的药代动力学用雄性小鼠常规方法测定(从略)。
2.3 三唑并哒嗪环的变换基于前述的喹啉环是选择性的必要结构和甲基吡唑的共有结构, 研制者固定这两个片段不变, 变换中央的并合杂环, 合成的代表性化合物的结构及其对c-Met激酶和对细胞中c-Met激酶的磷酸化的抑制活性列于表 1。
| Table 1 SAR of variation in the central core |
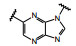
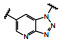
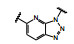
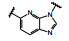
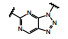
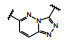
构效关系分析如下: ①化合物7和10的抑酶活性很强, 但10对细胞内抑制c-Met的磷酸化的活性表现一般; ② 8和11分别是去除7和10的N2原子, 导致活性降低, 提示N2对结合有贡献; ③ 9是将7的N8用CH置换, 即哌嗪并三唑换成吡啶并三唑, 活性显著下降, 抑制c-Met活性大于1 μmol·L-1, 提示N7之必需; ④化合物12是嘧啶并三唑, 即将7的N5移至N6, 抑酶活性明显下降; ⑤ 13是含有桥头的三唑母核, 抑制酶和细胞活性与10相近。
综上, 母核上氮原子的分布对活性影响显著, 推测是环上的电荷分布与c-Met酶的结合关系密切, 不止限于同Tyr1230的π-π叠合作用。7是活性最强的化合物。
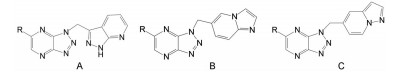
2.4 右侧喹啉环的变换这是一项重要的探索, 因为多家研制单位认为右侧的喹啉环是结合于铰链区并呈现选择性抑制c-Met的药效团, 如同化合物1~3和5之所示。和黄公司研究者变换该杂环, 并同时变换左侧咪唑环的取代基, 这应是探索超越(me-better) 的尝试。设计合成代表性化合物列于表 2。
| Table 2 SAR of variation in hinge binding moiety |
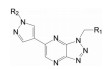
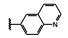
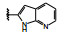
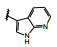
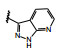
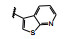
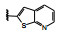
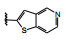
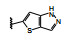
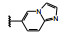
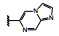
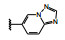
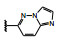
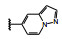
分析构效关系如下: 2-氮杂吲哚(14) 置换喹啉环, 活性显著下降, 而3-氮杂吲哚化合物15抑制酶和细胞增殖与7相当, 提示同样的片段连接的位置不同, 氮原子所处的空间位置则不同, 因而显著影响活性。15的2位引入氮原子成氮杂吲唑化合物16, 抑制酶和细胞活性与15相当。噻吩并吡啶环无论是2位连接(17) 或3位连接(18) 都降低活性, 但19移动了18的氮原子位置活性提高了17倍, 而噻吩并吡唑的20的活性更加提高, 可见这些片段上的氮原子的位置关乎与铰链的结合起到重要作用。19的结构中再加入1个氮原子, 20的活性仍很强。但最强的化合物21是吡唑并吡啶, 对c-Met和p-Met的抑制活性IC50分别为0.006和0.011 μmol·L-1, 抑制细胞增殖活性IC50 0.073 μmol·L-1。然而在21的杂环上任何位置添加氮原子(22~24、26、27) 都降低活性。左端的吡唑环尚需作深入研究, 但至此化合物21是活性最强的。
2.5 左端吡唑环的优化至此, 左端的吡唑环未作深入的构效关系考察。下一步是固定中央骨架为三唑并哌嗪, 右端选取活性较高的16、21和25的三个片段, 优化左端的杂芳环。
分析表 3的数据, 除了32B之外, 其余化合物对酶和细胞增殖都有较强的抑制活性, 尤其是28C、29A和29C活性很高, 31B虽然也是高活性抑制细胞增殖, 但肝微粒体代谢较快。进一步用荷瘤小鼠对高活性的15、16、21、28C、29A和29C化合物进行体内抑制磷酸化实验, 3 mg·kg-1灌胃6 h后测定抑制活性, 显示有中等或偏弱的活性。对16、21和28C测定药代动力学行为都有这样或那样的缺陷(数据从略), 因而尚需进一步结构优化。
| Table 3 SAR of compounds with the varied aromatic ring on the left hand and the selected hinge binding moieties |
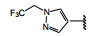
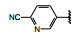
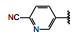
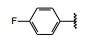
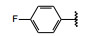
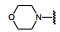
芳环侧链的α-亚甲基是容易氧化代谢的位点, 这或许是某些化合物体内不稳定的原因, α-位引入基团以增加位阻可降低氧化代谢。同时, 分析化合物6与c-Met复合物的结构生物学特征, 发现亚甲基临近有由Leu1157、Lys1110和Val1092组成的小疏水腔, 因而设计了α-位甲基取代的化合物, 并将对映体拆分成光活体, 如表 4所示。化合物33~36是两对对映异构体, 实验表明, S构型强于相应的R构型, 提示S-甲基有利于结合疏水腔中, 为优映体。这四个抑制c-Met酶活性与没有甲基取代的21和28C接近, 抑制增殖作用略强。但重要的是对于肝细胞生长因子(HGF) 诱导癌细胞增殖的抑制活性明显增强。
| Table 4 SAR of the compounds with an α-methyl substituent |
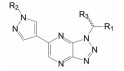
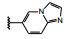
化合物33具有很好的体内外活性, 抑制细胞增殖活性强于化合物21大约12倍。小鼠实验比较了33与没有甲基取代的21的药代动力学, 如表 5所示。33的小鼠口服生物利用度虽然不如21, 但清除率、表观容积、半衰期、最大浓度和1~8 h的曲线下面积都优于21, 因而33在循环血中的持续暴露量高于21。33的小鼠的药效学也显著优于21 (数据从略)。在选择性上, 对274个激酶未显示脱靶现象(Jia H, Dai GX, Weng JY, et al. Discovery of (S)‑1-(1-(imidazo[1, 2‑a]pyridin-6-yl)ethyl)-6-(1-methyl‑1H‑pyrazol-4-yl)‑1H‑[1,2,3]triazolo[4, 5‑b]pyrazine(volitinib) as a highly potent and selective mesenchymal-epithelial transition factor (c-Met) inhibitor in clinical development for treatment of cancer. J Med Chem, 2014, 57: 7577-7589)。
| Table 5 Comparison of the pharmacokinetic profiles of compounds 21 and 33 |
为此, 确定了33作为候选化合物, 定名为赛沃替尼(savolitinib), 经临床前和临床研究表明, 用于治疗间质-上皮转化因子(c-Met) 外显子14跳变的晚期或转移性非小细胞肺癌患者, 经中国国家药品监督管理局(NMPA) 批准, 于2021年6月上市。赛沃替尼(商品名沃瑞沙) 成为我国首创的针对c-Met变异的非小细胞肺癌口服治疗药。
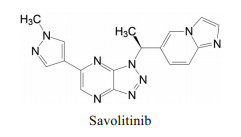
|
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多重性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
本品是上海和记黄埔研制的新药, 是针对间质-上皮转化因子(Met) 外显子14跳跃/变异的局部晚期或转移性非小细胞肺癌的治疗药, 成为世界上第三个针对Met靶标的药物, 本品似乎是跟随性药物, 其实是只有一定借鉴的创新型的项目。赛沃替尼的成功是用药物化学的方法结合结构生物学的特征实现的, 结构中的三个片段逐个进行优化, 从而突破了一些固有的认识。本品的研制过程填补了该领域的空白, 反映了我国当前创新药物的研究状态, 对于研发首创性(first in class) 药物也是一种蓄势。
(编者按)