 2022, Vol. 57
2022, Vol. 57



新药创制“以患者需求为核心, 以临床价值为导向”的提法, 似乎是理念的提升, 其实这并不是新的概念。2021年7月我国国家食品与药品监督管理局(NMPA) 公布了《临床价值为导向的抗肿瘤药物研发指导原则》, 这是自2005年《药品管理法》提出的“药物创新应以临床价值为导向”的具体要求, 也是落实2015年《国务院关于改革药品医疗器械审评审批制度的意见》文件精神。笔者相信这样的指导原则, 也将会扩展到其他治疗领域的药物。道理很简单, 新药创制的目的本应满足患者对无药可治或更加安全有效的药物需求。
科学的发展和医药生物技术的进步, 要求研制新药具有临床价值, 使患者受益, 这是用药最根本的目的和原则, 临床价值的内涵究竟包含哪些原则?笔者以为主要体现在以下内容: ①有效性: 新药的疗效要优于(或至少不劣于) 既有的药物疗法。②安全性: 新药的不良反应低于既有的药物, 如若安全性不如老药, 但必须在疗效方面更加突出, 权衡疗效/不良反应, 仍优于已有的疗法。③经济性: 包括两个方面, 一是疗效的提高与支付的研发成本应当合理, 也就是说, 新药价格不能太贵; 另一是新药治疗应使患者生活质量得到提高, 只延长患者的寿命而生活质量低下的药物治疗, 意义不大。由以上三个要素又派生出以下的药物特征: ①创新性, 主要体现在靶标和化学结构的新颖性。②可及性, 实现规模性制备和患者的经济承受力。③适宜性, 服用方便, 适宜的频次和疗程。新药创制的核心是患者的需求, 因而研制过程和候选药物须以实现临床价值为目标。研发链上各个环节都是临床价值的正向量。
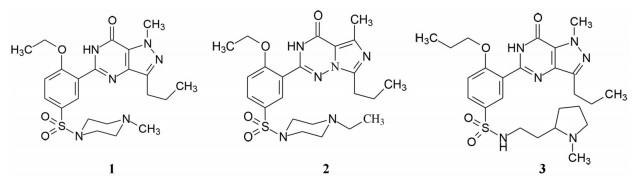
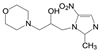
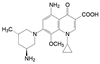
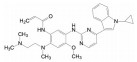
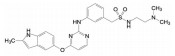
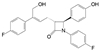
1 改革开放以来我国新药研制的梗概 1.1 前20年以仿制药物为主20世纪80年代到上世纪末, 制药企业和学/研界以仿制国外药物为主, 这对满足和更新各种疾病的药物疗法起到重大的推动作用。期间的创新药物虽有但很稀缺, 著名的青蒿素类抗疟药, 联苯双酯保肝降转氨酶药, 都是以天然产物为先导的成功药物。然而在抢仿过程中, 也出现了无序状态, 例如当时全国有100多家申报治疗男性勃起障碍用药的西地那非(1, sildenafil, 即伟哥, 其合成工艺简单, 收率高), 因处在专利期间被辉瑞提起诉讼, 结果是全体败诉。倘若当时有人仿创(me-too), 如同拜耳公司快速跟踪研制, 采用了骨架迁越和保持药效团的策略, 研制成功伐地那非(2, vardenafil), 或是韩国东亚药业研发的乌地那非(3, udenafil), 或许有中国自研的伟哥。进入WTO后, 开始由仿制转为创制。
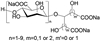
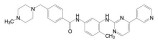
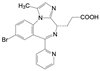
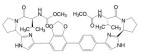
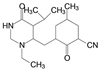
1.2 后20年以仿创性药物为主创新药物可分为两类: 首创性(first-in-class) 药物和仿创性(follow-on, me-too) 药物。首创性药物是全新的作用靶标/作用环节, 药物的结构和适应症都是全新的; 仿创性药物可认为是跟随性创制药, 是对已确证的靶标和已有研制的化合物(或药物) 的基础上进行的再创造。仿创药物无需发现和确证靶标的步骤, 但有效性/安全性上应优于首创药物, 后来居上。21世纪20年来的我国创新化学药物大都属于仿创性药物。表 1列出了2001~2021我国自行研制的1.1类化学药物。其中除双环醇、丁苯酞和槐定碱等天然产物属于首创性药物外, 绝大多数是仿创品种。从化学结构分析多数仿创药物含有首创药的结构特征或要素。
|
| Table 1 New chemical entities discovered in China and approved by NMPA during 2001-2021 |
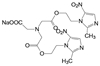
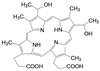
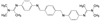
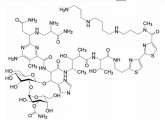


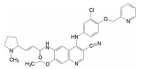
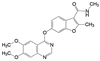
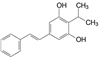
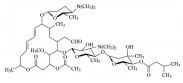



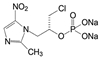

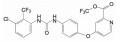
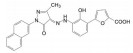
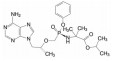
创新药物最珍贵的是首创性(first-in-class, FIC), 其成功造福于患者, 同时带来市场的回报。首创性药物的失败风险很大, 主要风险是靶标的非可药性(undruggability), 在概念验证或转化成实现临床价值有大概率的不成功性, 损失巨大, 尤其是研发后期的临床失败。例如目标为减肥药的蛋白酪氨酸磷酸酯酶1B (PTP1B) 抑制剂, 曾风靡一时, 体外活性达到纳摩尔水平, 但因体内达不到有效的浓度而无一成功; 阻止胆固醇吸收的胆固醇酯转运蛋白(CETP) 抑制剂, 数个候选化合物在III期临床中因出现安全性问题而全军覆没。首创药的风险不仅置前, 还贯穿于研发始终, 直至在真实世界的考量。
仿创药没有发现和确证靶标的投入和可药性风险, 但作为后继上市的药物, 除非品质显著优于先行者, 或适应症的特异性, 否则临床价值的实现和市场回报难以预期, 所以, me-too药的研制具有置后的风险, 而且竞争激烈。我国的制药工业体系已经形成, 研究单位和制药企业面临的是研制首创药, 还是守着仿创药的选择, 两种风险, 是进亦忧, 退亦忧, 但何以之选?
2.2 及早仿创, 避免无序竞争制药企业、生物技术公司、大学、研究院(所) 尽管价值取向不同, 但在创新药物的意义上仍有交集和相同处。笔者以为, 当前在首创、仿创方面二者应予并举, 并逐渐将重点移至首创性研究。在首创的内容方面, 宜不拘一格; 做仿创, 宜有的放矢, 周密的计划和论证, 聚焦于是否能够实现临床治疗价值, 这也是NMPA的批准原则, 避免一窝蜂似的挤在一个赛道上。
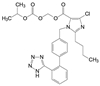
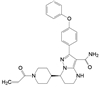
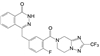
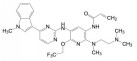
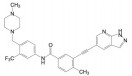
举个负面例子。FDA在2021年5月批准了美国安进公司首创的靶向KRASG12C小分子抑制剂索托拉西(4, sotorasib, AMG510), 治疗中晚期肺癌, 被认为是划时代的突破。一个月后, FDA又认定Mirati Therapeutics公司的KRASG12C抑制剂阿达拉西(5, adagrasib) 突破性疗法。此外, 国外处于临床研究的还有礼莱的LY3537982、诺华的TNO-155、加科思与艾伯维联合研发的JAB-3068、赛诺菲与Revolution联合研发的RMC-4630 (结构式从略) 等。这些药物的特点都在分子结构中含有迈克尔加成基团, 与KRAS的变构位点发生不可逆的共价键结合, 结合于KRASG12C变构区的Cys12 (亲电加成反应), 不可逆地锁定靶标于非活性状态。这是基于化学生物学研究发现了变构位点而成功转化的范例[1]。
我国业界以KRASG12C为靶标的新药研制, 出现过江之鲫般的仿创态势。有人统计, 我国有36家制药企业启动仿创, 申请了化合物专利75件, 大都是骨架、取代基或迈克尔片段的改动, 预计专利撞车不可避免, 被称为me-too药的修罗场(scene of bloody slaughter)。这种无序竞争的结果只能由NMPA操刀加以扬弃, 造成巨大的浪费。其他一些热门的靶标也呈现激烈的竞争, 例如PD-1、claudins蛋白家族抑制剂、CD47、SHP2、FGFR等重叠研制。仿创药物的路已经显露出日益狭窄的状态。
|
回顾我国研发新药历程, 20世纪80年代前, 由于不受专利约束, 主要是仿制国外的新药。直到21世纪初, 我国每年批准上市的1.1类化药还只是个位数。后来有了显著的发展, 国家批准了不少自研的1.1类化药(表 1), 这些新药的特点是: ①申报的候选药物95%是仿创药。②申报的每个品种至少有两个(重复研制), 少有像青蒿琥酯和蒿甲醚这样的全球性首创性药物。而国内发现的新靶标尚未得到国际的认可。③新药多集中在抗癌、抗病毒或抗感染药物。④不够重视在我国基数很大的罕见病药物和儿童用药的研究。⑤临床试验制定的标准偏低, 确定的临床价值缺少全面的证据。⑥临床试验的设计往往出于以临床注册作为目标, 而不是实现临床价值的追求。
3 创新药物以靶标为核心和以临床价值为导向的关系20世纪80年代开始的以靶标为核心的新药创制, 加速了新药的研发进程和药物治疗机制的阐明。随着经验的积累和激烈的竞争, 一些研制单位急于求成地将新药的创制理念逐渐异化为药物靶标/新分子实体(NME) 作为导向, 一定程度上忽视了满足患者需求和实现临床治疗价值的终极目标。认为有了新的靶标蛋白和高活性的化合物, 就有了卖点, 可以向前推进。由此, 似乎药物化学成为新药创制的主打。诚然, 在首创药物的研发链中, 发现和优化先导化合物, 最终获得候选化合物, 药物设计、合成和评价是重要的环节, 但这不是研发的全部, 更不能代替以临床价值为导向的策略和理念。以临床价值为导向, 旨在满足患者和医生的需求, 也是政府监管部门的诉求, 发达国家和我国审评新药的快速通道, 都体现了临床需求的特征, 在这当中, 并不强调或拘泥是否新靶标、NME, 只要在临床上有价值, 无论新化合物、老药、合并用药、新剂型甚至机制上不明确的药物(例如当年的青蒿素), 只要疗效好, 都是患者所需要的。以靶标为核心构建新分子目的是实现临床价值, 二者有交集, 但不能以前者代替后者。
满足患者需求和实现临床价值, 这是没有定式可遵循的, 内涵极其复杂, 一定程度上是摸着石头过河, 风险也大, 绝非新靶标/新分子那样简单。
4 新药研发实现临床价值的要素科学技术持续性发展是个长期的积累渐进过程, 虽然可以加大投入和提速, 但弯道超车的策略不可取, 应当借鉴一些国外的经验。实现临床价值的新药创制需建立在生物学基础研究上。为实现临床价值, 有人提出包含三个要素或环节, 即理解致病的分子机制; 把握患者的宏观表象和微观特征; 阐明药物的作用机制。
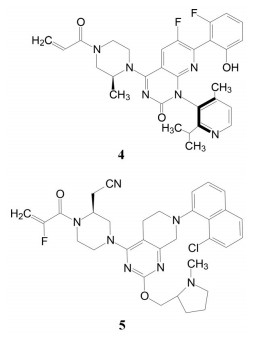
4.1 致病的分子机制上述的三个环节是创新药物实现临床价值的前提与保障, 三者是相互关联的。贯穿其中的是转化医学的实践, 即如何将生物学/医学基础研究的成果通过各种模型试验阐明疾病发生发展的分子机制, 从分析分子调控过程中找到治疗疾病的关键节点。例如20世纪末对自身免疫性疾病由于缺乏针对病因的治疗药物, 临床治疗大都是对症疗法。1993年美国NIH的生物学家O'shea等[2]发现了一个在免疫系统中起作用的激酶, 命名为JAK-3激酶, JAK-3只在淋巴细胞表达, 与白介素-2 (IL-2) 受体的γ链结合, 调节信号转导。IL-2是T淋巴细胞的关键生长因子, 具有排斥异体器官移植的作用。选择性抑制JAK-3激酶, 可以抑制或调节机体的免疫功能。辉瑞化学家Changelian等[3]立即着手研究针对JAK-3这个新靶标蛋白的药物, 辉瑞与NIH之间签订了合作协议, 构建了多种测定JAK-3的体内外活性的模型。由化学生物学开始, 通过化合物的普筛, 找到分子探针研究靶标的生理功能, 反复探索JAK-3与免疫功能的因果关系, 以确证是否是药物靶标, 同时也是寻找活性化合物的过程。经过化学生物学和药物化学以及临床研究, 研制出托伐替尼(6, tofacitinib) 于2012年FDA批准上市, 成为首创的口服治疗类风湿性关节炎、牛皮癣和炎性大肠炎的药物[4]。
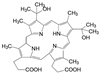
艾伯维与罗氏公司合作研发的抗癌药维特克拉(7, venetoclax), 于2016年4月经FDA批准上市, 治疗携带17p删除/突变的复发性/难治性慢性淋巴细胞白血病, 这是全球第一个针对蛋白-蛋白相互作用的小分子抑制剂, 研发历程长达20年。该药物对发病的分子机制和靶标的确定经历了曲折过程: 由开始以BCL-XL作为靶标, 中间改换为作用于BCL-XL和BCL-2双靶标, 最后才聚焦于BCL-2靶标, 彰显了首创性药物的曲折与艰辛, 围绕着靶标与疾病的因果关系展开了反复的验证, 甚至在确定了候选化合物乃至进入临床试验, 仍然对于靶标与候选物作了更换, 提示药物靶标的验证贯穿于研发的全过程, 始终存在风险[5]。
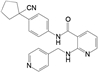
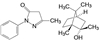
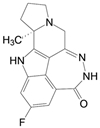
由疾病的分子机制转化到新药创制, 是一个研判过程。我国应世生物科技公司在转化医学上颇有建树。公司与上海交通大学瑞金医院合作, 发现在KRASG12C抑制剂作用下, 狡黠的癌细胞通过诱导活化黏着斑激酶(FAK) 使自己产生耐药性, 从而抵消了KRASG12C抑制剂的疗效。他们用建立的CDX和PDX模型证实FAK抑制剂(8, N10018) 和KRASG12C抑制剂索拖拉西(4) 联合用药, 能实现协同的抗癌作用, 降低了耐药反应, 使抗癌作用更加持久。进一步的机制研究发现, 异常激活的FAK-YAP通路是KRASG12C抑制剂产生耐药作用的重要原因[6]。8已在美国、澳大利亚和中国开展三项临床研究。应世生物显示了高效的转化医学和全球临床研发的能力。
|
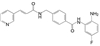
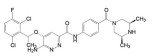
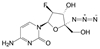
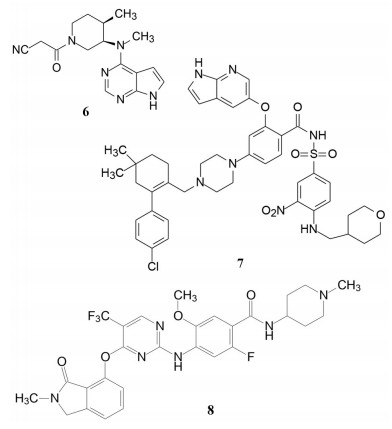
罕见病镰刀状红细胞病(SCD) 治疗药沃噻洛托(10, voxelotor) 的创制, 体现了从解析疾病的分子机制, 经分析患者的病理特征, 最后基于药物的作用机制研制出特效药物, 于2019年批准上市治疗SCD。SCD是由于血红蛋白β链的Glu6变异为Val6, 在低氧条件下发生四聚化, 导致红细胞变形成镰刀状, 载氧量降低使毛细血管堵塞, 造成贫血和器官衰竭。沃噻洛托含有的芳香醛基是从先导物5-羟甲基呋喃醛(9, 5-HMF) 优化得到的。5-HMF与突变的血红蛋白复合物的晶体结合模式提示, 醛基与突变体的N末端Val的氨基缩合成西佛碱, 呈1∶1的化学计量结合, 阻断了血红蛋白的四聚化, 减少了镰刀细胞的生成。10醛基邻位的羟基对于调节醛基的亲电性和降低脱靶性有重要影响[7]。
|
基于患者的疾病特征发明新药或药物疗法, 经典的范例是发现幽门螺杆菌(HP), 改变了传统治疗胃炎/胃溃疡的疗法。20世纪80年代以前治疗胃炎/胃溃疡的主要手段是胃黏膜保护剂(氢氧化铝或铋制剂) 和H2受体阻滞剂(如西咪替丁等)。1979年病理学家Warran经胃镜/活检在胃炎患者胃窦黏膜组织中发现一种弯曲状细菌, 细菌所在之处细胞黏膜发生炎症, 1982年他与消化科医生Marshall合作, 分离并培养出HP菌, 证明了HP感染是胃病的主要病原体, 二人的重要发现获得了2005年诺贝尔奖。
由于发现了HP是胃炎/十二指肠溃疡的病原菌, 使得诊断和治疗手段有显著的进步, 例如用同位素标记的尿素测定HP尿素酶的活力作为诊断方法, 用治本药物(甲硝唑+阿奇霉素等抗生素) 和治标药物(铋/铝制剂/替普瑞酮+替丁类药物) 的三联/四联疗法, 成为治疗HP引起胃炎的标准疗法。
既往对患者的临床表现, 多基于症状、生化指标、组织切片和影像特征加以描述和判断, 虽然有更多的客观指标, 但仍属于宏观的表象。从药物创新的层面审视, 并没有在分子水平(基因或蛋白) 揭示疾病特征的内在联系。如今分子生物学的发展, 得以在微观水平作生物学分析, 例如基因缺失与变异、蛋白质组学、转录组学、表观遗传等技术, 分析患者的临床表现, 从多样本和大数据分析归纳基因-蛋白(酶、受体、通道、信号分子等) 与疾病的关系, 为创制新药和药物疗法提供新的科学依据。因而洞察疾病的宏观/微观特征是上述发病的分子机制的延续和深化, 为创制新药和疗法提供可行的科学路径。
例如抗肿瘤药物的研究更能体现上述的关联性。人体恶性肿瘤病有100多种(不包括亚型), 作为复杂性疾病, 干预单一的靶标或生物标志物往往难以控制肿瘤的发展。这是因为恶性肿瘤有许多异常的功能, 概括起来有10个共同的特征, 其中任何一个特征都可以成为肿瘤细胞逃逸药物治疗的栈道或陈仓。这十大特征是: 肿瘤细胞无控制的分裂; 诱导血管生成; 激活肿瘤细胞的浸润和迁移; 逃逸抑制剂的阻断; 逃避免疫蛋白的杀伤; 促进炎症因子生成; 阻止程序性死亡(细胞凋亡); 失控的能量需求; 基因的变异和不稳定性; 启动持续的增殖信号。所以即使特异性地抑制某个靶标或关键的生物标志物也未必能够达到完全的治疗效果。
BRAF是人类最重要的原癌基因之一, 位于人类7号染色体上, 编码RAF家族丝氨酸/苏氨酸蛋白激酶, BRAF蛋白在调节MAPK/ERK信号通路中起介导作用, 影响细胞分裂、分化和分泌。大约8%的肿瘤发生BRAF突变, 突变位点是BRAFV600E, 主要发生于黑色素瘤、结肠癌和甲状腺癌中。该突变导致下游MEK-ERK信号通路持续激活, 促进了肿瘤的生长增殖和侵袭转移, 所以, BRAFV600E是抗黑色素瘤等的重要作用靶标。
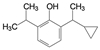
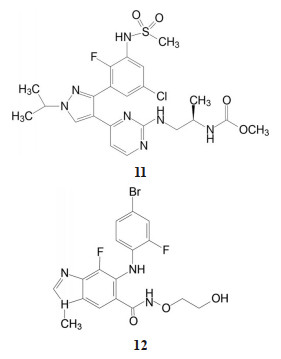
2018年6月FDA批准Array BioPharma公司研发的两个激酶抑制剂康奈非尼(11, Encorafenib, 商品名Braftovi)[8]和比美替尼(12, Binimetinib, 商品名Mektovi)[9], 联合应用治疗无法切除或转移的BRAF V600E或V600K突变阳性的黑色素瘤。这是一个典型的基于疾病分子机制而成功设计的治疗药。
|
康奈非尼是BRAF激酶抑制剂, 阻断了MAPK/ERK信号通路; 比美替尼是MEK的选择性抑制剂, MEK是促进肿瘤的MAPK途径中的中枢激酶, 所以康奈非尼和比美替尼联合用药协同治疗BRAF V600E或V600K突变阳性黑素瘤治疗。
然而在临床治疗BRAFV600E发生变异的黑色素瘤和结直肠癌中, 发现有显著的差异, 肠癌患者联合用药后, 效果仍显不足, 是因为上述联合治疗后, 癌细胞启动了表皮细胞生长因子(EGFR) 的信号通路, 分子生物学显示异常活跃, 以逃逸康奈非尼+比美替尼的“制裁”。为此, 将EGFR抑制剂西妥昔单抗(cetuximab) 与康奈非尼或比美替尼二联合用, 或三联疗法, 疗效显著提高, 从而FDA于2021年9月批准用康奈非尼+西妥昔单抗二联组合治疗BRAF V600E发生变异的结直肠癌。
4.3 药物的作用机制20世纪80年代以前的药物大都是在细胞或整体动物的表型模型上创制的, 诞生了许多优质药物。证明在临床的真实世界应用是安全有效的。当阐明了疾病的分子机制和药物的作用机制(靶标或信号通路), 对既有的药物(或活性物质) 的应用有了更深刻或广泛的认识, 不仅提供了新的思路, 还扩大了药物的适应症。举例如下。
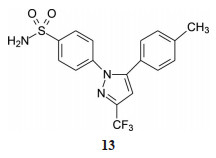
4.3.1 前列腺素和环氧合酶抑制剂20世纪70年代发现人体前列腺素及其功能(发现者J Vane获得了1982年诺奖), 打开了镇痛和抗炎的新领域, 并对后来发现的环氧合酶1和2的功能、对非甾体抗炎药(NSAID) 和COX2选择性抑制剂(例如首创药物塞来昔布13, celecoxib) 的抗炎镇痛有了新的认识, 从分子水平和机制上解析了抗炎镇痛-胃肠道不良反应-心血管事件之间的关系。还确定了阿司匹林在防止血小板聚集和血栓形成的新用途[10]。
|
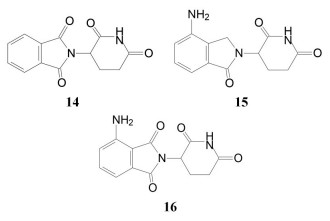
20世纪60年代一度用作孕妇的镇静药沙利度胺(14, thalidomide), 因引起严重的致畸作用而臭名昭著, 但后来发现沙利度胺具有免疫调节及抗炎作用, 以及抑制血管生成及抗肿瘤作用, 还发现抑制某些细胞因子的分泌如血管内皮生长因子(EGF) 和成纤维细胞因子(FGF), 这些生长因子都是血管生成的刺激剂, 这些蛋白与相应受体作特异性结合, 刺激信号转导, 引起内皮细胞的增殖。沙利度胺干预该路径的血管生成。重要的是14作为手性分子, 致畸作用与抗肿瘤活性是立体选择性的, 因而可完全避免该不良反应。由沙利度胺研制的来那度胺(15, lenalidomide) 和泊马度胺(16, pomalidomide) 分别于2008和2014年被FDA批准上市, 治疗多发性骨髓瘤, 延误了近半个世纪。
另外, “度胺”类药物还发现其他特性。基础研究揭示泛素-蛋白酶体系统(ubiquitin-proteasome system, UPS) 是重要的科学发现(三位发现者获得2004年诺贝尔化学奖), UPS的生理功能是清除有害或错误蛋白, 通过酶促的生物化学反应, 将蛋白降解成小肽, 彻底摧毁蛋白的功能。这个成果经转化医学, 开拓出降解致病蛋白的新的治疗理念, 诞生了蛋白降解靶向嵌合体(PROTAC) 技术。PROTAC是双功能小分子, 一端是结合于被降解蛋白的配体, 另一端是泛素连接酶E3的配体, 经连接基共价连接成单一分子。PROTAC的功能是将靶蛋白与E3连接酶募集在一起, 将靶标蛋白泛素化, 旋即被蛋白酶体降解, 使靶标蛋白彻底失活。
|
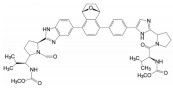
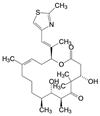
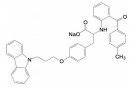
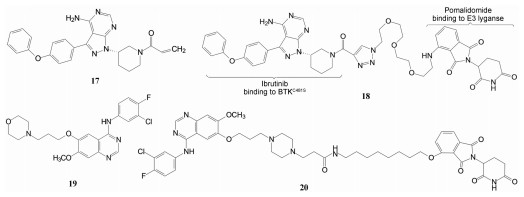
上述的泊马度胺(16) 是泛素E3连接酶cereblon的配体, 连接成PROTAC分子。可将Bruton酪氨酸激酶(BTK) 泛素化, 泛素化的BTK被蛋白酶体降解而失活, 因而泊马度胺可作为研制PROTAC药物的元件, 以治疗套细胞淋巴瘤和慢性淋巴白血病。尤其可对一线用药的伊鲁替尼(17, ibrutinib) 已发生耐药性的患者是有潜在的价值, 这是因为BTK的Cys481变异为Ser481, 17结构中的迈克尔基团因Cys481不存在亲核性巯基而失去作用, PROTAC (18) 作为BTK降解剂, 可望克服耐药性。18中的泊马度胺片段和伊鲁替尼类似物(迈克尔基团已无用武之地, 恰作为连接的位点) 分别是E3连接酶cereblon和靶标BTK激酶的配体, 18用于治疗对伊鲁替尼发生耐药的套细胞淋巴瘤和慢性淋巴白血病[11]。
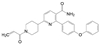
第一代EGFR激酶抑制剂吉非替尼(19) 用于治疗非小细胞肺癌已近20年, 治疗中会因酶的突变, 逃逸抑制而失效, Cheng等[12]将吉非替尼6位侧链的吗啉环(不参与同激酶结合的助溶基团) 变为哌嗪作为设计PROTAC的连接位点, 用沙利度胺作为E3连接酶cereblon的招募基, 通过系统的构效关系研究, 优化出PROTQC分子MS154 (20), 对突变型的EGFRL858R结合的Ki值为3.8 nmol·L-1, 20选择性地对突变的EGFR降解, 而对野生型作用很弱, 反映了PROTAC分子作为事件驱动的过程与占据驱动过程所产生的后果是不同的。
|
溴域4 (BRD4) 在多种肿瘤高表达, 是肿瘤治疗的靶标, 噻吩三唑并二氮䓬化合物(如OTX015, 21) 是BRD4的配基, Lu等[13]将泊马度胺经聚乙二醇连接, 化合物22 (代号ARV-825) 可诱导蛋白酶体选择性地降解BRD4蛋白, IC50 < 1 nmol·L-1。22可降低Brukitt淋巴瘤细胞中C-myc水平, 阻断细胞增殖, 并诱导凋亡。体外实验加入来那度胺可阻断22的降解作用, 是因为游离的来那度胺分子竞争性地结合cereblon, 阻止了BRD4的泛素化作用。
以上含有泊马度胺类的PROTAC例子说明, 发掘已有的药物的新作用机制可以用于创制新的治疗领域, 其实PROTAC结构中结合靶标蛋白的配基可以广泛地借鉴已有药物或活性分子。
例如为了治疗去势前列腺癌而又耐受雄受体(AR) 拮抗剂的患者, Han等[14, 15]研究AR的PROTAC降解剂, 以“鲁胺”类AR拮抗剂为结合AR的配基, 分析了恩杂鲁胺(23) 和其他AR拮抗剂与雄受体的共晶结构, 发现连接胺酰基的苯环未同受体接触, 因而以此作为连接位点连接招募E3连接酶VHL的配体, 对连接基作多轮优化, 得到了24, 对高表达AR的LNCaP细胞的IC50 = 2 nmol·L-1, 而对照的AR拮抗剂恩杂鲁胺的IC50 = 150 nmol·L-1。
4.3.3 胰高血糖素样肽-1的长效化治疗慢性病需长期用药, 提高药物的代谢稳定性, 减少用药的频次或方便的服药方式也是实现临床价值的有效途径。丹麦的诺和诺德公司在蛋白质类药物的长效化和口服化的首创性引人注目, 他们的降糖和减肥药的GLP-1类药物已占据全球市场很高的份额。
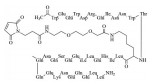
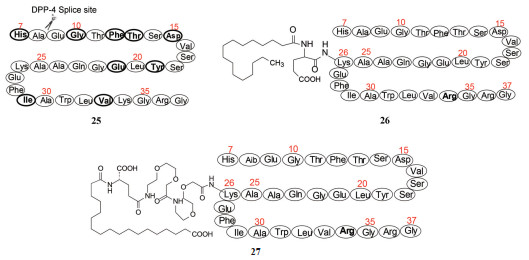
胰高血糖素样肽-1 (25, GLP-1) 是肠内分泌的肽类激素, 产生于人小肠黏膜的L细胞。GLP-1具有多种生理功能: 刺激胰岛的β细胞分泌胰岛素, 抑制高血糖素的释放, 延缓胃的排空, 以及抑制食欲等, 这些效应都是葡萄糖依赖性的, 因而GLP-1是生理性降低血液中葡萄糖的内源性调节物质。所以, 只要维持一定水平的GLP-1, 理论上应是控制2型糖尿病的有效途径。GLP-1发现于1984年, 是含有30或31个氨基酸的肽类激素[16]。
人体内激素的共同特点, 是一旦分泌并履行生理功能后, 就被迅速代谢失活, 以维持内环境的稳态。GLP-1也具有快速被降解的性质, 是被二肽基肽酶(DPP-4) 水解, 半衰期低于2 min, 酶的剪切位点为Ala8-Glu9, 生成的代谢产物与受体的亲和力很低, 因而失去活性。诺和诺德的研究者用丙氨酸扫描确定了同受体结合的重要氨基酸残基(25中粗体所示), 反衬出可以修饰或变换的位点。
经过多轮的构效关系研究最终优化出利拉鲁肽(26, liraglutide), 其结构是将Lys34变换成Arg34, 在Lys26的ξ-氨基经谷氨酸连接基, 再与十六烷酸连接的GLP-1修饰肽。
白蛋白是多功能的转运蛋白, 可运载内源性物质和药物。被转运的分子上若连接长脂肪(酸) 链可提高与白蛋白的结合能力, 从而延长在血浆中的存留时间并减低被肾脏的清除。经临床前实验和临床研究, 证明利拉鲁肽具有与GLP-1相同的生理功能, 在血浆中利拉鲁肽发生自缔合(self association) 并与白蛋白发生疏水性结合, 显著提高了对DPP-4和内切酶的稳定性, 半衰期t1/2 11~15 h, 患者每日用特制注射器自行皮下注射一次, 可控制2型糖尿病患者的血糖, 成为第一个改构的人GLP-1的降血糖药物, 美国FDA于2010年批准上市[17]。GLP-1对DPP4的不稳定性是由于Ala8-Glu9是酶的剪切位点, 为了提高对DPP4的稳定性, 在研制利拉鲁肽的构效关系基础上, 将利拉鲁肽的Ala8换成α-氨基异丁酸Aib8, 以降低DPP4的识别和剪切, 变换并增长连接于Lys26的脂肪链, 又研制出新一代的修饰肽索马鲁肽(27, semaglutide)[18]。表 2比较了索马鲁肽(27) 与利拉鲁肽(26) 的药代动力学性质, 索马鲁肽在血浆的半衰期是利拉鲁肽的4倍, 生物利用度高达94%, 清除率比利拉鲁肽低一半。
| Table 2 Pharmacokinetic evaluation in Gőttingen mini-pigs following administration of semaglutide (27, 2 nmol·kg-1 i.v. or 2 nmol·kg-1 s.c.) and liraglutide (26, 0.5 nmol·kg-1 i.v. or 1.0 nmol·kg-1 s.c.) |
索马鲁肽于2017年批准上市, 作为治疗糖尿病和减肥药物, 每周注射或口服用药1次, 实现了长效和方便应用的临床目标。2019年索马鲁肽的销售额达17亿美元, 2020年上半年15亿美元, 已超过了利拉鲁肽。
4.3.4 西地那非潜在的新用途西地那非(1, sildenafil) 可谓是传奇式药物。最初是辉瑞公司针对PDE5靶标研发治疗冠心病和肺动脉高压药物, 在临床II期阶段, 对有限数量的心绞痛患者进行实验治疗观察, 结果没有达到预期的效果而终止了心血管病的临床研究。以Osterloh为首的临床医生在分析归纳西地那非的临床效果时, 意外地发现男性受试者服药后生殖器出现勃起现象, 勃起程度与服用剂量呈相关性。这个发现促使辉瑞公司改变了该项目的适应症研究, 在1994年对12名有勃起障碍的患者进行了Ⅱ期临床研究, 结果10名患者的勃起有显著增强。经过Ⅲ期临床研究, 证实西地那非确实有促进男性勃起的功能, 于1998年FDA批准作为促进勃起的药物上市, 后来临床证实对治疗肺动脉高压也有明确的疗效[19]。
|
后继的基础研究和临床研究提示, 西地那非与降低阿尔茨海默病风险可能存在因果关系, 认为在阿尔茨海默病患者中一氧化氮/环磷酸鸟苷(cGMP) 水平降低, 是由于降解cGMP的磷酸二酯酶5 (PDE5) 上调的缘故, 西地那非抑制PDE5, 增加cGMP水平。西地那非还抑制β-分泌酶1的表达和淀粉样蛋白-β (Aβ) 的生成, 上调抗氧化酶, 并诱导线粒体K通道开放等。西地那非还可增强神经发生, 同时抑制神经细胞凋亡和炎症[20]。
分子生物学和计算生物学为老药新用提供了有力的支撑, 扩展了已有药物的用途。Cheng (程雄飞) 等用计算生物学整合了阿尔茨海默病的生物学和遗传学数据, 构建了该疾病13个特征性内表型网络(endophenotype network), 绘制成包含有35万种人类蛋白-蛋白相互作用的网络图, 再用1 600种已知药物拟合网络接近度的分值, 高分值药物预示能与阿尔茨海默病的多种靶标发生相互作用。发现西地那非是得分最高的一种药物, 结合临床前研究西地那非可以显著改善认知和记忆, 提示西地那非可能影响阿尔茨海默病。为了验证西地那非与阿尔茨海默病的相关性, 还分析了美国700多万人的保险理赔数据, 发现使用西地那非的男性随访6年后阿尔茨海默病确诊风险降低了73%。细胞生物学研究表明, 用患者的人类小胶质细胞和神经元(可诱导多能干细胞) 进行体外实验, 西地那非可增加神经轴突生长, 并降低tau蛋白的异常磷酸化, 说明西地那非在降低阿尔茨海默病风险有潜在的用途[21]。
4.3.5 魔高一尺道高一丈的Bcr-Abl激酶抑制剂诺华公司2001年上市的伊马替尼(28, imatimib) 是作用于Bcr-Abl激酶、治疗慢性粒细胞白血病的首创性药物, 成为开辟靶向药物新领域的标志。持续用药使靶标Bcr-Abl发生变异, 激酶的门户氨基酸残基The315变异为Ile315, 使得28难以与之结合而无效。2007年上市的尼洛替尼(29, nilotinib) 是诺华研制克服耐药的第二代产品, 是通过构效关系研究得到的。29与28的结构主要区别是两个苯环间的胺酰键-HN-CO-逆向为酰胺键-CO-NH-, 以及右侧苯环连接较大的取代基团, 结构生物学表明, 酰胺基变向的29更适配于同突变型激酶发生氢键结合, 29对转染Bcr-Abl变异的Ba/F3细胞活性比28提高了26倍[22]。

|
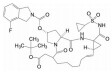
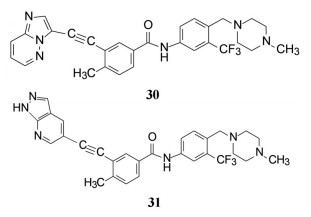
2012年Ariad公司上市的帕纳替尼(30, ponatinib) 是日本武田公司基于AblT315I变异的微观结构“量体裁衣”式设计出来的。Thr315变异为Ile, 残基侧链的CH2OH变成CH(Me)Et, 失去了OH, 使嘧啶与苯环之间的-NH-不能形成氢键, 28的-NH-已无所用, 而且突变后的Ile体积和疏水性都大于Thr, 空间变小, 30用炔键连接两个芳环, -C≡C-体积减小了位阻并有利于疏水结合, 因而对AblT315I变异的瘤株显著提高了活性, 为第三代治疗慢粒白血病的药物[23]。我国亚盛医药研制的GZD824 (31) 也是克服耐药的新一代药物, 临床研究对耐药的慢性髓性白血病已显示有良好治疗效果[24]。
|
新药创制是以生物学基础研究驱动, 患者需求为核心, 临床价值为导向, 转化医学为杠杆的复杂的科学技术体系, 作为引擎的基础生物学研究成果, 通过化学生物学、药物化学、药理学(包括药代动力学和毒理学等)、转化医学和临床医学的接力整合, 实现新药创制的知识价值链。构建新的分子结构(NME) 的药物化学是其中的重要环节, 但不是核心, 实现药物的临床价值、满足患者的需求则是终极目标, 现时形势要求药化工作者应突破固有的靶标-设计-合成-评价的模式, 从大视野的角度, 上溯关注基础研究及其成果, 及时向创制新药的方向转化; 向下游关注临床要求, 结合疾病的宏观和微观特征, 落实转化过程的具体内容, 这对于首创性药物的研发是非常关键的环节。
| [1] |
Ostrem JM, Peters U, Sos ML, et al. K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions[J]. Nature, 2013, 503: 548-551. DOI:10.1038/nature12796 |
| [2] |
Kawamura M, McVicar DW, Johnston JA, et al. Molecular cloning of L-JAK, a Janus family protein-tyrosine kinase expressed in natural killer cells and activated leukocytes[J]. Proc Natl Acad Sci U S A, 1994, 91: 6374-6378. DOI:10.1073/pnas.91.14.6374 |
| [3] |
Changelian PS, Flanagan ME, Kent CR, et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor[J]. Science, 2003, 302: 875-878. DOI:10.1126/science.1087061 |
| [4] |
Flanagan ME, Blumenkopf TA, Brissette WH, et al. Discovery of CP-690, 550:a potent and selective Janus kinase (JAK) inhibitor for the treatment of autoimmune diseases and organ transplant rejection[J]. J Med Chem, 2010, 53: 8468-8484. DOI:10.1021/jm1004286 |
| [5] |
Park CM, Bruncko M, Adickes J, et al. Discovery of an orally bioavailable small molecule inhibitor of prosurvival B-cell lymphoma 2 proteins[J]. J Med Chem, 2008, 51: 6902-6915. DOI:10.1021/jm800669s |
| [6] |
Zhang BY, Zhang Y, Zhang JW, et al. Focal adhesion kinase (FAK) inhibition synergizes with KRAS G12C inhibitors in treating cancer through the regulation of the FAK-YAP signaling[J]. Adv Sci, 2021, 8: 2100250. DOI:10.1002/advs.202100250 |
| [7] |
Metcalf B, Chuang C, Dufu K, et al. Discovery of GBT440, an orally bioavailable Rstate stabilizer of sickle cell hemoglobin[J]. ACS Med Chem Lett, 2017, 8: 321-326. DOI:10.1021/acsmedchemlett.6b00491 |
| [8] |
Li Z, Jiang K, Zhu X, et al. Encorafenib (LGX818), a potent BRAF inhibitor, induces senescence accompanied by autophagy in BRAFV600E melanoma cells[J]. Cancer Lett, 2016, 370: 332-344. DOI:10.1016/j.canlet.2015.11.015 |
| [9] |
Serra V, Eichhorn PJA, García-García C, et al. RSK3/4 mediate resistance to PI3K pathway inhibitors in breast cancer[J]. J Clin Invest, 2013, 123: 2551-2563. DOI:10.1172/JCI66343 |
| [10] |
Guo ZR. Innovation of anti-inflammatory drugs-strategy of moderate inhibition of cyclooxygenases[J]. Acta Pharm Sin (药学学报), 2005, 40: 967-969. |
| [11] |
Sun YH, Zhao XW, Ding N, et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies[J]. Cell Res, 2018, 28: 779-781. DOI:10.1038/s41422-018-0055-1 |
| [12] |
Cheng M, Yu XF, Lu K, et al. Discovery of potent and selective epidermal growth factor receptor (EGFR) bifunctional small-molecule degraders[J]. J Med Chem, 2020, 63: 1216-1232. DOI:10.1021/acs.jmedchem.9b01566 |
| [13] |
Lu J, Qian Y, Altieri M, et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4[J]. Chem Biol, 2015, 22: 755-763. DOI:10.1016/j.chembiol.2015.05.009 |
| [14] |
Han X, Wang C, Qin C, et al. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer[J]. J Med Chem, 2019, 62: 941-964. DOI:10.1021/acs.jmedchem.8b01631 |
| [15] |
Han X, Zhao LJ, Xiang WG, et al. Discovery of highly potent and efficient PROTAC degraders of androgen receptor (AR) by employing weak binding affinity VHL E3 ligase ligands[J]. J Med Chem, 2019, 62: 11218-11231. DOI:10.1021/acs.jmedchem.9b01393 |
| [16] |
Thorens B. Expression cloning of the pancreatic β-cell receptor for the gluco-incretin hormone glucagon-like peptide 1[J]. Proc Natl Acad Sci U S A, 1992, 89: 8641-8645. DOI:10.1073/pnas.89.18.8641 |
| [17] |
Knudsen LB, Nielsen PF, Huusfeldt PO, et al. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration[J]. J Med Chem, 2000, 43: 1664-1669. DOI:10.1021/jm9909645 |
| [18] |
Lau J, Bloch P, Schäffer L, et al. Discovery of the once-weekly glucagon-like peptide 1(GLP-1) analogue semaglutide[J]. J Med Chem, 2015, 58: 7370-7380. DOI:10.1021/acs.jmedchem.5b00726 |
| [19] |
Ghofrani HA, Osterloh IH, Grimminger F. Sildenafil: from angina to erectile dysfunction to pulmonary hypertension and beyond[J]. Nat Rev Drug Discov, 2006, 5: 689-702. DOI:10.1038/nrd2030 |
| [20] |
Sanders O. Sildenafil for the treatment of Alzheimer's disease: a systematic review[J]. J Alzheimers Dis Rep, 2020, 4: 91-106. DOI:10.3233/ADR-200166 |
| [21] |
Fang J, Zhang P, Zhou Y, et al. Endophenotype-based in silico network medicine discovery combined with insurance record data mining identifies sildenafil as a candidate drug for Alzheimer's disease[J]. Nat Aging, 2021. DOI:10.1038/s43587-021-00138-z |
| [22] |
Manley P, Stiefl N, Cowan-Jacob S, et al. Structural resemblances and comparisons of the relative pharmacological properties of imatinib and nilotinib[J]. Bioorg Med Chem, 2010, 18: 6977-6986. DOI:10.1016/j.bmc.2010.08.026 |
| [23] |
Huang WS, Metcalf CA, Sundaramoorthi R, et al. Discovery of 3-[2-(imidazo[1, 2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl)-methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a potent, orally active pan-inhibitor of breakpoint cluster region-abelson (BCR-ABL) kinase including the T315I gatekeeper mutant[J]. J Med Chem, 2010, 53: 4701-4719. DOI:10.1021/jm100395q |
| [24] |
Ren XM, Pan XF, Zhang Z, et al. Identification of GZD824 as an orally bioavailable inhibitor that targets phosphorylated and nonphosphorylated breakpoint cluster region-abelson (Bcr-Abl) kinase and overcomes clinically acquired mutation-induced resistance against imatinihem[J]. J Med Chem, 2013, 56: 879-894. DOI:10.1021/jm301581y |