 2020, Vol. 55
2020, Vol. 55



2. 江西中医药大学药学院, 江西 南昌 330004
2. School of Pharmacy, Jiangxi University of Traditional Chinese Medicine, Nanchang 330004, China
二硫代氨基甲酸酯被认为是最简单的有机硫化合物, 含有此类结构单元的衍生物在化工和药学领域应用广泛[1]。二硫代氨基甲酸酯在20世纪被用作杀虫剂, 此后由于其良好的金属结合能力而引起了许多药物化学家们的关注。近年来, 除了金属结合能力之外, 还探索了二硫代氨基甲酸酯衍生物的多种生物活性, 包括抗氧化[2]、抗病毒[3]、抗菌[4]、抗肿瘤[5]和抗阿尔茨海默病等活性。本文将简单介绍二硫代氨基甲酸酯及其衍生物的结构和合成, 重点描述文献中报道的二硫代氨基甲酸酯及其衍生物的生物活性, 希望为二硫代氨基甲酸酯结构在药物化学中的应用提供参考。
1 二硫代氨基甲酸酯及其衍生物的结构二硫代氨基甲酸酯(1)的结构可以看作是硫代氨基甲酸酯(2)结构中的氧原子被硫原子取代而形成的, 其中, 氨基-1-碳二硫代酸(3)是二硫代氨基甲酸酯的药效基团, 对二硫代氨基甲酸酯结构上的R、R1、R2进行改造, 则会得到一系列的衍生物, 不同的衍生物在不同的介质中, 其稳定性是不同的, 比如, 在酸性介质中, N原子上的R1或R2被取代的单烷基二硫代氨基甲酸酯比R1和R2同时被取代的双烷基二硫代氨基甲酸酯更稳定, 这是由于第二个烷基会对C-N键产生空间作用力, 而且会产生诱导效应使得N原子的电子云密度增加, 影响了S-H键的极性, 最终导致双烷基二硫代氨基甲酸酯稳定性降低[6]。
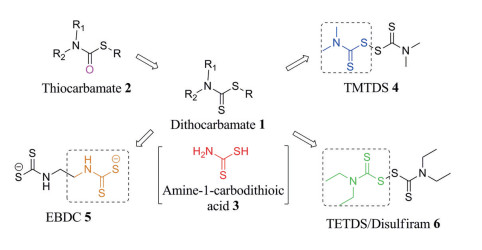
|
据报道, 对二硫代氨基甲酸酯进行结构修饰, 得到的第一个衍生物是二硫化四甲基秋兰姆(4), 属于一种农业杀菌剂, 通常称为福美双[7]。除此之外, 还有乙撑双二硫代氨基甲酸酯(5)类杀菌剂, 包括代森钠、代森锌和代森锰锌等, 他们都属于首类广谱叶用杀菌剂, 已被用于防治植物病害50多年, 目前依然在使用[8]。二硫化四乙基秋兰姆(6), 俗称戒酒硫, 自20世纪40年代以来一直在临床上用于慢性酒精中毒治疗[9]。戒酒硫于1881年首次合成, 且用于加速橡胶硫化。20世纪30年代, 戒酒硫用于作疥疮和杀螨剂[10]。在1948年, 它开始用于治疗慢性酒精中毒, 作为戒酒治疗[11]。
2 二硫代氨基甲酸酯及其衍生物的合成二硫代氨基甲酸酯衍生物作为一类重要的含硫化合物, 其合成方法的研究受到了长期广泛的关注, 并且取得了较大进展。据报道, 在早期, 二硫代氨基甲酸酯的合成是通过胺与有毒且不易得的试剂, 如硫代光气反应而制得的(图 1a)[12]。目前, 二硫代氨基甲酸酯衍生物常用的合成方法是:在氢氧化钠或氢氧化钾存在的强碱性条件下, 二硫化碳与伯胺或仲胺反应生成对应的二硫代氨基甲酸盐, 再与卤代烷反应, 生成二硫代氨基甲酸酯(图 1b)[13]。由于反应环境为强碱性, 所以反应底物中不能含有对碱敏感的基团, 同时反应中间体需要分离, 工艺复杂, 反应时间较长。

|
Figure 1 Synthesis routes of dithiocarbamates under different reaction conditions |
后来, Azizi[14]和Ranu[15]两支团队选择了不同的绿色溶剂作为溶剂和催化剂, 成功地合成了二硫代氨基甲酸酯衍生物, 其中, Azizi团队选择以水为溶剂和催化剂(图 1c), Ranu团队选择以离子液体([pmIm]Br, 1-methyl-3-propylimidazolium bromide)为溶剂和催化剂(图 1d), 是二硫代氨基甲酸酯衍生物绿色有机合成的较好范例。但研究发现, 以水为溶剂时, 反应时间较长, 产物需要大量溶剂来萃取; 以离子液体为溶剂时, 虽然反应时间较短, 但产物分离也需大量溶剂来萃取, 所以并不是完全意义上的绿色合成。
近年来, 无溶剂有机合成逐渐成为有机合成领域的研究热点之一。相对于传统的溶液反应来说, 无溶剂有机反应具有绿色、经济、高效、高选择、易分离纯化产物及反应条件温和等优点[16]。在此背景下, Guo等[17]在无溶剂条件下, 采用“一锅法”成功合成了二硫代氨基甲酸酯衍生物。实验发现, 即使在无溶剂和无催化剂的条件下, 该反应也能较快地完成, 而且产率较高, 是一种合成二硫代氨基甲酸酯衍生物较好的方法(图 1e)。
3 二硫代氨基甲酸酯及其衍生物的生物活性在过去的十几年中, 二硫代氨基甲酸酯已经成为了药物化学中一个重要的药效团和结构。药物化学家们合成了一系列的二硫代氨基甲酸酯衍生物, 并对其进行了各种生物活性的评价。下面将从二硫代氨基甲酸酯衍生物的抗氧化活性、抗病毒活性、抗菌活性、抗肿瘤和抗阿尔茨海默病等方面进行阐述。
3.1 抗氧化活性众所周知, 抗氧化剂在保护生物体方面具有重大作用, 它能够使细胞免受氧化应激反应引起的损伤。一般活性氧(ROS, reactive oxygen species)存在就会引起氧化应激反应, 其中, ROS包括超氧自由基阴离子、羟基自由基和过氧化氢等。ROS通常会对人体的蛋白质、脂质和DNA造成损害, 从而加速衰老, 引起炎症、癌症、心血管疾病和神经退行性疾病等。所以, 开展相关合成与天然药物的抗氧化性研究是必不可少的。
研究表明, 二硫代氨基甲酸酯及其衍生物具有良好的抗氧化活性, 在药物的设计中被多次应用。例如, Onwudiwe等[18]设计并合成了N-乙基-N-苯基二硫代氨基甲酸酯的Cu (Ⅱ)配合物(7), 考察了Cu (Ⅱ)配合物的抗氧化活性。结果表明, 该化合物在500 μg·mL-1时具有75%的自由基清除活性。Oladipo等[19]研究了镍(Ⅱ)-和铜(Ⅱ)-N, N′-二芳基甲酰胺二硫代氨基甲酸酯复合物的抗氧化活性, 实验结果显示, 化合物8a和8b具有最佳的抗氧化活性。
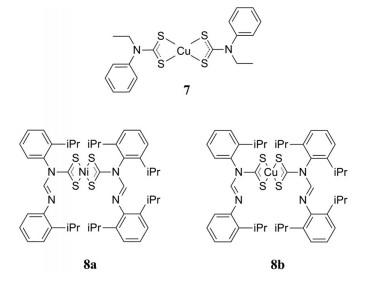
|
人类鼻病毒(HRVs)最早发现于20世纪50年代, 导致一半以上的人患上感冒。然而, 由于人类鼻病毒的易发性和多变性, 到目前为止, 预防或治疗人类鼻病毒的特定药物还没有出现。有研究发现, 吡咯烷二硫代氨基甲酸酯(9)具有非常有效的抗人类鼻病毒作用, 而且对几种细胞系的HRVs亚型都显示了很好的抑制作用[20]。
人类免疫缺陷病毒(HIV)属于一种慢病毒, 能够攻击人体免疫系统, 感染人体免疫系统细胞, 使得人体免疫系统失去抵抗能力, 导致艾滋病的发生。He等[21]设计合成了一系列环状二硫代氨基甲酸酯结构, 并评价了其对于HIV-1包膜糖蛋白gp41靶点的融合抑制活性。最有效的化合物10表现出抑制gp41的六螺旋束结构(gp41 6-HB)的形成和HIV-1的复制, IC50值和EC50值分别为1.8和0.3 μmol·L-1, 分子对接结果表明化合物10的多芳香环骨架能够与关键残基K574产生相互作用。
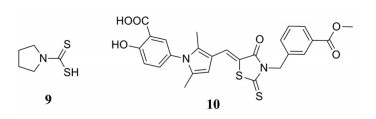
|
康唑类药物是目前临床上最有效的抗真菌药物, 然而该类药物存在一定的毒性和耐药性, 在一定程度上限制了其临床应用。2014年, Zou等[22]基于康唑类化合物的结构特点, 设计合成了一系列含有二硫代氨基甲酸酯结构的衍生物11, 并评价了它们的体外抗真菌活性。实验结果显示, 大多数化合物的抗念珠菌活性均高于对照药物氟康唑和酮康唑, 结果还表明, 二硫代氨基甲酸酯结构的引入能够有效地增加衍生物对真菌的抑制活性, 特别是二硫代氨基甲酸酯结构中N原子上的取代基R1、R2为不同苯基取代的哌嗪基时(化合物12系列), 抗真菌活性最好。Ke等[23]在进行化合物结构设计时, 在保留三氮唑基团的基础上, 在侧链上引入二硫代氨基甲酸酯结构, 设计并合成了一系列衍生物13, 希望获得活性更好的抗真菌候选药物。活性评价结果表明, 该系列衍生物均具有一定的抗真菌活性。
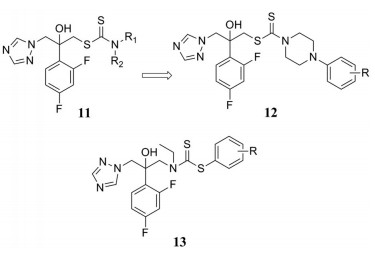
|
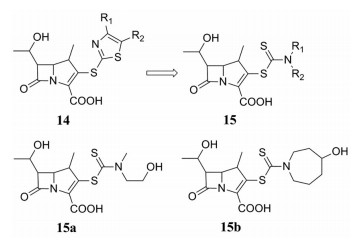
β-内酰胺类抗生素杀菌力强、毒性低, 在临床上应用广泛, 特别是3-位上有噻唑环的β-内酰胺抗菌剂。Ohtake课题组[24]用二硫代氨基甲酸酯结构替换化合物14中的噻唑结构, 设计并合成了一系列含有β-内酰胺结构的二硫代氨基甲酸酯衍生物15, 并评价该类化合物的抗菌活性。结果表明, 该类化合物均具有很好的抗菌活性, 其中化合物15a和15b的活性最好, 对金葡菌抑制的MIC值分别为0.025和0.012 μg·mL-1, 明显优于万古霉素(MIC = 0.39 μg·mL-1), 而化合物15a和15b对表皮葡萄球菌抑制的MIC值分别为6.25和3.13 μg·mL-1。多年来, β-内酰胺类药物的广泛使用使得许多细菌对其产生了耐药性, 所以新型抗菌素的研发成为热点。Wang等[25]发现二硫代氨基甲酸酯结构的引入不但使化合物具有较强的抗菌活性, 而且能够通过抑制β-内酰胺酶的活性来对抗细菌的耐药性。
|
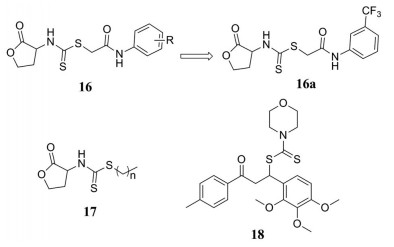
酰化高丝氨酸内酯类(AHLs)化合物是革兰阳性菌群体感应系统中最重要的一类信号分子, 能够调控许多生理特性的表达, 是目前研究的热点。Liu团队[26]设计并合成了两个系列含二硫代氨基甲酸酯结构的AHLs类化合物16和17, 并对其体外抗菌活性进行了评价。抗菌活性结果显示, 化合物16系列对革兰阳性菌具有很强的活性, 特别是化合物16a对枯草芽孢杆菌的抑制作用最强, MIC50值可达1.443 μg·mL-1, 而令人遗憾的是, 化合物17系列对革兰阳性菌有较弱的抑制活性。
查尔酮结构简单, 是许多生物活性物质的重要组成部分, 包括降压、抗肿瘤、抗炎和抗氧化等。Ayman等[27]设计并合成了一系列新型含有查尔酮结构的二硫代氨基甲酸酯衍生物, 对某些革兰阴性菌进行了抗菌筛选。结果发现, 大多数合成化合物都表现出较好的抗菌活性, 其中, 化合物18被认为是最佳候选药物, 而且在一定程度上能够对抗细菌的耐药性。
|
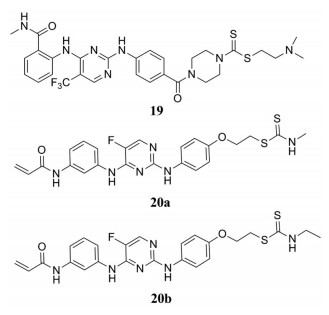
癌症仍然是全世界死亡的主要原因之一。然而, 传统的化疗对肿瘤细胞缺乏选择性, 在杀灭癌细胞的同时也对机体正常细胞造成损伤, 因此, 近年来具有特定靶标的小分子抗癌化合物成为研究的重点, 此种化合物具有较好的选择性和有效性。其中粘附斑激酶(FAK)、布鲁顿酪氨酸激酶(BTK)等非受体酪氨酸激酶常常被选作抗癌药物的靶点, 对应的酪氨酸激酶抑制剂是治疗癌症的有效策略。研究发现, 二苯基胺嘧啶和丙烯酰胺是酪氨酸激酶抑制剂的药效基团。Yin团队[28]采用分子拼合的方法, 设计了一系列含有哌嗪二硫代氨基甲酸酯结构的2, 4-二芳基氨基嘧啶类化合物, 筛选出最佳FAK抑制剂。实验结果显示, 化合物19对HCT116、PC-3、U87-MG和MCF-7细胞株具有良好的抗增殖作用, 还表现出显著的FAK抑制活性(IC50 = 0.07 nmol·L-1)。除此之外, 该团队还改变2, 4-二芳基氨基嘧啶在二硫代氨基甲酸酯结构上的取代位置, 将其与二硫代氨基甲酸酯的S原子通过合适连接结构进行连接, 得到一系列化合物, 筛选出了最佳BTK抑制剂20a、20b[29]。
|
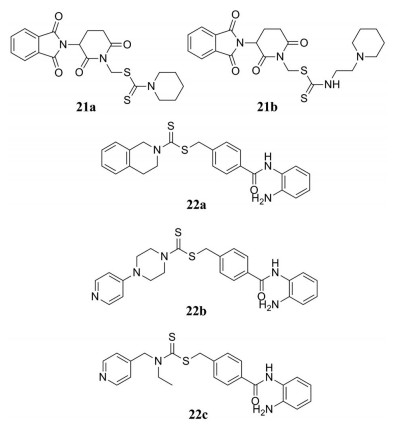
据报道, 组蛋白去乙酰化酶(HDACs)会在不同类型的癌细胞中过度表达, 过度表达的HDACs催化组蛋白, 去除乙酰基, 导致染色质浓缩, 抑癌基因下调, 而HDAC抑制剂能够选择性地诱导癌细胞周期阻滞、分化和凋亡, 因此, HDAC抑制剂已经成为治疗癌症的有效策略之一。Zahran等[30]设计并合成了一系列含沙利度胺结构的二硫代氨基甲酸酯衍生物, 并进行了体外抗肿瘤活性的研究。实验发现, 化合物21a和化合物21b对HepG2、HCT-116细胞株及HDAC具有一定的抑制作用。Xie等[31]以二硫代氨基甲酸酯为基础结构, 设计合成了一系列新型2-氨基苯甲酰胺类化合物, 筛选其作为HDAC抑制剂的活性大小。实验结果表明, 二硫代氨基甲酸酯结构的引入可有效提高HDAC的抑制活性和抗肿瘤活性, 其中, 化合物22a~22c对多种人肿瘤细胞株具有很强的抗增殖活性, 是此类型的最佳的HDAC抑制剂。
|
金属酶碳酸酐酶(CAs)参与许多重要的细胞过程, 如呼吸、pH稳态、电解质分泌、骨吸收以及生物合成过程, 除此之外, 研究发现, 它在肿瘤生长和转移中具有一定的作用, 其中两种同工酶(CA-I和CA-II)是体内最丰富的CAs, 已成为许多药物的靶点。Altintop等[32]设计并合成了一系列含有亚甲二氧基苯基结构的二硫代氨基甲酸酯衍生物23, 并检测了其对A549人肺腺癌、C6大鼠胶质瘤细胞以及对人CA-I (hCA-I)和人CA-II (hCA-II)的抑制作用。检测结果显示, 化合物23a和23b具有最佳的抑制hCA-I和hCA-II的活性, 其中, 对hCA-I抑制的IC50值分别为0.346和0.288 nmol·L-1, 对hCA-II抑制的IC50值分别为0.287和0.338 nmol·L-1。
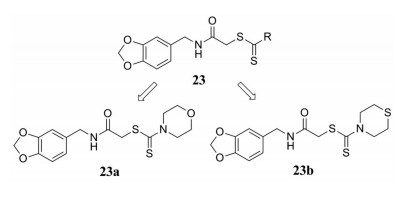
|
以1, 3-二苯基丙烯酮为基本骨架结构的查尔酮是天然产物中的重要药效团, 也是合成黄酮和异黄酮类化合物的重要前体, 具有广泛的生物学活性。Qian等[33]设计并合成了一系列含有查尔酮结构的二硫代氨基甲酸酯衍生物, 并评价了它们作为抗增殖剂和微管蛋白聚集抑制剂的活性。其中化合物24不仅表现出最强的体外抗肿瘤活性, 其IC50值为0.04 μmol·L-1, 也表现出对微管蛋白聚集有较强的抑制作用, 其IC50值为6.8 μmol·L-1。Liu团队[34]设计并合成了一系列新型二硫代氨基甲酸酯-查尔酮类化合物, 并对3种癌细胞株(EC-109、SK-N-SH和MGC-803)的抗增殖活性进行了评价。实验结果表明, 大多数化合物对3种癌细胞株都表现出中等到强的抑制活性, 特别是化合物25a和25b对SK-N-SH具有良好的生长抑制作用。
γ-丁烯内酯又叫2(5H)-呋喃酮, 是一类广泛存在于天然产物中的重要化学活性单元, 大部分该类化合物都表现出抗真菌、抗细菌、抗病毒和抗肿瘤等生物活性。Liu团队[35]设计并合成了3个系列的含丁烯内酯的二硫代氨基甲酸酯衍生物, 并对其体外抗肿瘤活性进行了考察。活性评价结果显示, 化合物26具有广谱抗癌活性, 对5株人癌细胞(EC-9706、HeLa、PC-3、SPCA1、MCF-7)抑制的IC50值均小于30 μmol·L-1。构效关系考察结果说明, γ-丁烯内酯C-3位置上二硫代氨基甲酸酯侧链的引入对抗肿瘤活性至关重要。
黄酮类化合物属于植物次生代谢物, 具有抗氧化、抗肿瘤、抗真菌等广泛的药理活性, 是天然产物研究的重点。Huang等[36]设计并合成了4个系列的含有黄酮母核的二硫代氨基甲酸酯衍生物27~30, 探索它们之间的构效关系, 并进行抗肿瘤活性的评价。构效关系研究表明, 二硫代氨基甲酸酯结构通过亚甲基连接在类黄酮3位的化合物28系列的抗肿瘤活性明显优于其他系列化合物。
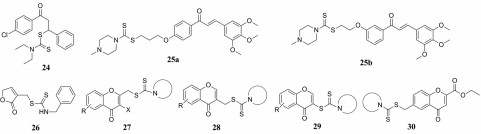
|
三唑衍生物具有抗炎、抗肿瘤、抗菌、止痛等广泛的生物活性, 并能在一定程度上预防或对抗细菌耐药性。2013年, Liu团队[37]报道了一系列含1, 2, 3-三唑结构的二硫代氨基甲酸酯衍生物的合成及对4种人肿瘤细胞株的抗肿瘤活性的测试结果。结果发现, 化合物31系列表现出广谱的抗肿瘤活性, 化合物31a和31b对MGC-803、MCF-7、PC-3和EC-109四种细胞株抑制的IC50值的范围分别在0.73~11.61和0.49~12.45 μmol·L-1。同年, 该团队在上述化合物的基础上进一步设计, 将1, 2, 3-三唑-二硫代氨基甲酸酯结构和尿素结构相结合得到一系列新型化合物32, 其中化合物32a和32b具有广谱抗癌活性, 而且对MGC-803癌细胞抑制作用非常明显[38]。2014年, Liu团队[39]还在化合物31的基础上, 用香豆素替代了化合物31中的取代苯基结构, 并评价了该类化合物抑制组蛋白去甲基化酶的活性。研究结果显示, 香豆素在不同的取代位置对化合物活性影响很大, 其中化合物33对组蛋白去甲基化酶的抑制活性最好, 其IC50值为0.39 μmol·L-1。
喹唑啉酮结构具有抗癌、抗菌、抗病毒等多种药理活性, 尤其是4(3H)喹唑啉酮类化合物的结构修饰与改造引起了广泛的关注。Cao团队[40]将二硫代氨基甲酸酯结构与4(3H)-喹唑啉酮结构进行拼合, 合成了一系列新型二硫代氨基甲酸酯衍生物34, 并进行体外活性评价。实验结果表明, 化合物34a对白血病K562细胞株的抑制活性最强, 其IC50为0.5 μmol·L-1。后来, 该团队又对化合物34系列进行了两次改造, 得到两个系列化合物35和36, 第一次是对二硫代氨基甲酸酯部分进行改造, 得到最佳化合物35a; 第二次是将4(3H)-喹唑啉酮结构改造成2, 4-二氨基喹唑林结构, 得到最佳化合物36a~36c[41, 42]。
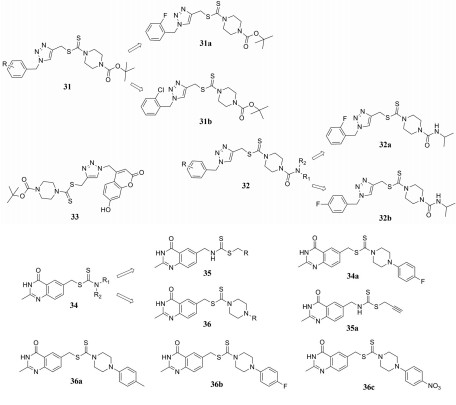
|
芒柄黄花素是一种具有生物活性的异黄酮, 广泛存在于豆科, 据报道, 其具有许多有效的药理活性, 包括抗氧化、抗病毒、抗肿瘤、抗高血压、抗菌、抗血管生成等作用。近年来, 芒柄黄花素结构被用于抗肿瘤药物的设计, 例如Liu团队[43]将芒柄黄花素结构与二硫代氨基甲酸酯结构进行拼合, 设计合成了一系列化合物37, 并对3种癌细胞株(MGC-803、EC-109、PC-3)的抗增殖活性进行了评价。评价结果显示, 化合物37a具有最佳的抗肿瘤活性, 对PC-3抑制的IC50值可达1.97 μmol·L-1。
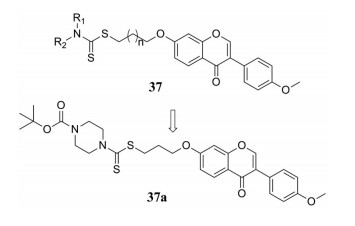
|
研究表明, 青蒿素及其衍生物都表现出不同程度的抗肿瘤活性。例如, 二氢青蒿素(DHA)可选择性诱导胃癌细胞凋亡。世界卫生组织曾推荐使用对人体无害的青蒿素及其衍生物来治疗肿瘤, 或在它们的结构中引入其他抗肿瘤活性基团, 这将是治疗肿瘤有效的策略之一。Yu研究小组[44]合成了一系列二氢青蒿素-哌嗪二硫代氨基甲酸衍生物, 并考察了它们在体外抗肝细胞瘤的活性。其中, 化合物38对SMMC-7721细胞株具有最佳的抑制活性, 其IC50为0.002 5 μmol·L-1。
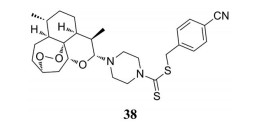
|
阿尔茨海默病(AD)是一种与年龄相关的神经退行性疾病, 其特点为记忆力减退、语言能力下降和认知障碍等[45]。如今, 全球有近3 600万人正在遭受阿尔茨海默病的影响, 而且到2050年, 预计这一人数将会增加3倍。因此, AD已经成为严重的世界公共卫生问题, 急需解决。AD起病隐匿, 发病机制尚未阐明, 许多因素被认为与AD的发生和发展有关[46]。
在AD机制阐述中, 经典的“胆碱能假说”已被研究人员广泛接受, AD患者的乙酰胆碱(ACh)水平降低会导致记忆缺陷和认知障碍, 减少ACh水解有利于AD的治疗, 所以, 乙酰胆碱酯酶(AChE)抑制剂对于治疗AD具有一定的价值[47]。Altintop等[48]设计并合成了N, N-取代的二硫代氨基甲酸酯衍生物, 并对其抗AChE活性进行了评价, 实验结果表明, 二硫代氨基甲酸酯结构具有抗AChE活性, 且含有哌嗪的化合物39的IC50值为0.53 μmol·L-1, 具有最强的AChE抑制活性。除此之外, Altintop等[49]还报道了一些具有二硫代氨基甲酸酯结构的吡唑啉类化合物, 它们可作为新型AChE抑制剂, 其中化合物40是最有效的AChE抑制剂, IC50值为0.72 μg·mL-1。
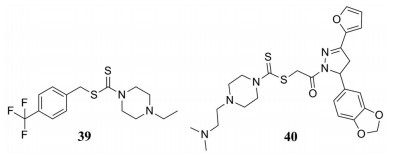
|
AChE的晶体结构表明, 它有一个深而窄的峡谷, 谷底是催化活性位点(CAS), 在谷的入口处, 有一个调节位点称为外周阴离子位点(PAS)。本课题组研究发现, 二硫代氨基甲酸酯结构能够与AChE的CAS位点结合, 达到抑制AChE活性的目的[50]。2018年, 本课题组将天然产物香豆素结构和二硫代氨基甲酸酯结构进行拼合, 得到一系列具有多靶点的新型香豆素-二硫代氨基甲酸酯衍生物41, 并进行了相关活性评价。结果发现, 大多数化合物对AChE具有较强的抑制作用和良好的选择性, 并能够抑制自身诱导的β淀粉样蛋白(Aβ)的聚集, 其中, 化合物41a对AChE的抑制能力最高, IC50可达0.027 μmol·L-1, 而且能够很好地抑制自身诱导的Aβ聚集。分子对接结果表明, 化合物41a能够同时作用于AChE的CAS和PAS位点, 为双位点AChE抑制剂。此外, 还具有特定的金属螯合性能和良好的血脑屏障通透性[51]。同年, 本课题组将香豆素结构上的取代基和二硫代氨基甲酸酯结构的N上的取代基进行调整, 希望得到具有AChE和单胺氧化酶B (MAO-B)双靶点抑制活性的二硫代氨基甲酸酯衍生物, 最终设计合成了系列化合物42, 并进行了相关活性评价。研究结果表明, 化合物42a为一系列中活性最强的AChE抑制剂, 活性优于对照药物多奈哌齐, 化合物42b具有最平衡的AChE/MAO-B双靶点抑制活性[52]。2019年, 本课题组将色满酮与二硫代氨基甲酸酯结构拼合起来, 设计并合成了一系列新型多功能AChE抑制剂。实验结果表明, 大多数化合物对AChE具有很强的选择性抑制作用, 而且对自身诱导和AChE诱导的Aβ聚集也具有很强的抑制作用, 其中, 化合物43抑制AChE的IC50值为0.01 μmol·L-1 [53]。
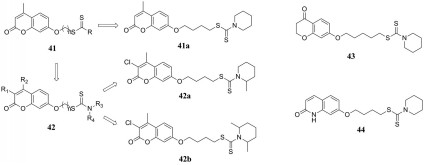
|
2020年, 本课题组设计并合成了一系列含有二硫代氨基甲酸酯结构的喹啉酮类化合物, 并对其相关生物活性进行了检测。结果显示, 大多数化合物对AChE具有较高的选择性抑制活性, 其中, 化合物44具有最强的AChE抑制活性, 同时也能够有效地抑制AChE诱导的Aβ聚集和自身诱导的Aβ聚集[54]。
“氧化应激假说”也是普遍接受的AD发病机制的阐述之一。研究发现, 内源性防御氧化应激受核因子E2相关因子2 (Nrf2)控制, 通过上调Nrf2信号来平衡氧化应激的影响已被证明在涉及氧化应激的各种疾病模型中是有效的, 其中包括AD。因此, 设计合成小分子激活剂刺激Nrf2信号上调细胞内源性防御机制是治疗AD的有效策略之一。Liddell团队[55]发现吡咯烷二硫代氨基甲酸酯(9)是星形胶质细胞中Nrf2信号的有效激活剂, 并证明Nrf2在吡咯烷二硫代氨基甲酸酯介导的抗氧化应激保护中具有关键作用。此外, 实验还证明, β-淀粉样蛋白的存在会增强吡咯烷二硫代氨基甲酸酯介导的内源性保护作用。而β-淀粉样蛋白沉积是AD的病理学特征之一, 由此可见, 吡咯烷二硫代氨基甲酸酯可能是AD背景下有效的Nrf2激活剂, 在AD治疗方面具有重要作用。
4 总结与展望本文对二硫代氨基甲酸酯及其衍生物的结构、合成和生物活性进行了综述。从文中内容可以看出, 随着现代化工合成技术的不断进步, 要求的不断提高, 二硫代氨基甲酸酯及其衍生物的新的合成方法不断涌现, 反应过程更加绿色环保。二硫代氨基甲酸酯也因其独特分子结构, 使得它的衍生物具有广泛的生物活性, 已经成为大量学者研究的热点之一, 特别是近几年发现的抗阿尔茨海默症的活性, 进一步拓展了其在药物化学研究中的应用。相信在不久的将来, 具有更多更高活性的二硫代氨基甲酸酯衍生物将会被不断发现与开发, 其应用也必将进一步丰富和加强, 在有机合成和药物化学领域占有越来越重要的地位。
作者贡献:郭杰负责全文的撰写以及图片等的制作; 张志鹏、程茂军参与文章相关资料的调研; 刘婧、周维新参与文章的校正与修改; 本文通讯作者为谢赛赛, 提供文章的撰写思路, 负责文章的审阅与修改
利益冲突:所有作者无利益冲突
| [1] |
Li RD, Wang YQ, Ge ZM, et al. Progress in the studies on synthesis and biological properties of dithiocarbamates[J]. Chin J Org Chem (有机化学), 2015, 35: 1805-1819. |
| [2] |
Schreck R, Meier B, Mannel DN, et al. Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells[J]. J Exp Med, 1992, 175: 1181-1194. |
| [3] |
Gaudernak E, Seipelt J, Triendl A, et al. Antiviral effects of pyrrolidine dithiocarbamate on human rhinoviruses[J]. J Virol, 2002, 76: 6004-6015. |
| [4] |
Menezes DC, Vieira FT, de Lima GM, et al. Tin (Ⅳ) complexes of pyrrolidine dithiocarbamate:synthesis, characterisation and antifungal activity[J]. Eur J Med Chem, 2005, 40: 1277-1282. |
| [5] |
Amir KM, Khan SZ, Hayat F, et al. Anticancer activity, DNA-binding and DNA-denaturing aptitude of palladium (II) dithiocarbamates[J]. Inorg Chim Acta, 2016, 451: 31-40. |
| [6] |
Sun HY, Sun HS, Liu MZ, et al. Lead optimization and antiproliferative activity using a new dithiocarbamates substructure[J]. Chin J Org Chem (有机化学), 2018, 38: 2067-2075. |
| [7] |
Board PW, Holland RV, Elbourne RGP. The effect of sulphur-containing fungicides on the corrosion of plain cans of fruit[J]. J Sci Food Agric, 1967, 18: 232-236. |
| [8] |
Singh DK. Bioremediation of hazardous ethylenebisdithio-carbamate (EBDC) fungicides[J]. Prog Inorg Chem, 2002, 36: 573-582. |
| [9] |
Chen D, Dou QP. New uses for old copper-binding drugs:converting the pro-angiogenic copper to a specific cancer cell death inducer[J]. Expert Opin Ther Target, 2008, 12: 739-748. |
| [10] |
Eneanya DI, Bianchine JR, Duran DO, et al. The actions of metabolic fate of disulfiram[J]. Annu Rev Pharmacol Toxicol, 1981, 21: 575-596. |
| [11] |
Hald J, Jacobsen E. A drug sensitizing the organism to ethyl alcohol[J]. Lancet, 1948, 2: 1001-1004. |
| [12] |
Kanchi S, Singh P, Bisetty K. Dithiocarbamates as hazardous remediation agent:a critical review on progress in environmental chemistry for inorganic species studies of 20th century[J]. Arabian J Chem, 2014, 7: 11-25. |
| [13] |
Lieber E, Orlowski RO. Notes-hydrazinolysis of 1-(alkyldithioate)-piperidine[J]. J Org Chem, 1957, 22: 88-89. |
| [14] |
Azizi N, Aryanasab F, Torkiyan L, et al. One-pot synthesis of dithiocarbamates accelerated in water[J]. J Org Chem, 2006, 71: 3634-3635. |
| [15] |
Ranu BC, Adak L, Chattopadhyay K. Hydroxyapatite-supported palladium-catalyzed efficient synthesis of (E)-2-alkene-4-ynecarboxylic esters[J]. J Org Chem, 2008, 73: 5609-5612. |
| [16] |
Yu FC, Yan SJ, Lin J. Application of solvent-free reaction in synthesis of heterocyclic compounds[J]. Chin J Org Chem (有机化学), 2010, 30: 1421-1430. |
| [17] |
Guo SR, Yuan YQ, Zhang CN. Highly efficient catalyst-free one-pot synthesis of dithiocarbamates under solvent-free conditions[J]. Chin J Org Chem (有机化学), 2012, 32: 907-914. |
| [18] |
Onwudiwe DC, Ekennia AC. Synthesis, characterization, thermal, antimicrobial and antioxidant studies of some transition metal dithiocarbamates[J]. Res Chem Intermediat, 2017, 43: 1465-1485. |
| [19] |
Oladipo SD, Omondi B, Mocktar C. Synthesis and structural studies of nickel (II)-and copper (II)-N, N'-diarylformamidine dithiocarbamate complexes as antimicrobial and antioxidant agents[J]. Polyhedron, 2019, 170: 712-722. |
| [20] |
Yokokawa F, Nilar S, Noble CG, et al. Discovery of potent non-nucleoside inhibitors of dengue viral RNA-dependent RNA polymerase from a fragment hit using structure-based drug design[J]. J Med Chem, 2016, 59: 3935-395. |
| [21] |
He XY, Lu L, Qiu JY, et al. Small molecule fusion inhibitors:design, synthesis and biological evaluation of (Z)-3-(5-(3-benzyl-4-oxo-2-thioxothiazol-idinylidene)methyl)-N-(3-carboxy-4-hydroxy)phenyl-2, 5-dimethylpyrroles and related derivatives targeting HIV-1 gp41[J]. Bioorg Med Chem, 2013, 21: 7539-7548. |
| [22] |
Zou Y, Yu SC, Li RW, et al. Synthesis, antifungal activities and molecular docking studies of novel 2-(2, 4-difluorophenyl)-2-hydroxy-3-(1H-1, 2, 4-triazol-1-yl)propyldithiocarbamates[J]. Eur J Med Chem, 2014, 74: 366-374. |
| [23] |
Ke XF, Zhang ZQ, Wu MC, et al. Synthesis and antifungal activities of novel triazole derivatives with dithiocarbamates side chain[J]. Pharm Care Res (药学服务与研究), 2016, 16: 11-14. |
| [24] |
Ohtake N, Imamura H, Jona H, et al. Novel dithiocarbamate carbapenems with anti-MRSA activity[J]. Bioorg Med Chem, 1998, 6: 1089-1101. |
| [25] |
Wang MM, Chu WC, Yang Y, et al. Dithiocarbamates:efficient metallo-β-lactamase inhibitors with good antibacterial activity when combined with meropenem[J]. Bioorg Med Chem Lett, 2018, 28: 3436-3440. |
| [26] |
Ren JL, Zhang E, Ye XW, et al. Design, synthesis and antibacterial evaluation of novel AHL analogues[J]. Bioorg Med Chem Lett, 2013, 23: 4154-4156. |
| [27] |
Ayman M, El-Messery SM, Habib EE, et al. Targeting microbial resistance:synthesis, antibacterial evaluation, DNA binding and modeling study of new chalcone-based dithiocarbamate derivatives[J]. Bioorg Chem, 2019, 85: 282-292. |
| [28] |
Su Y, Li RD, Ning XL, et al. Discovery of 2, 4-diarylaminopy-rimidine derivatives bearing dithiocarbamate moiety as novel FAK inhibitors with antitumor and anti-angiogenesis activities[J]. Eur J Med Chem, 2019, 177: 32-46. |
| [29] |
Zhai Z, Li RD, Bai XY, et al. Design, synthesis and biological evaluation of novel dithiocarbamate-substituted diphenylamino-pyrimidine derivatives as BTK inhibitors[J]. Bioorg Med Chem, 2019, 27: 4124-4142. |
| [30] |
Zahran MAH, Gamal-Eldeen AM, El-Hussieny EA, et al. Thalidomide dithiocarbamate and dithioate derivatives induce apoptosis through inhibition of histone deacetylases and induction of caspases[J]. J Gen Eng Biol, 2014, 12: 65-70. |
| [31] |
Xie R, Li Y, Tang PW, et al. Design, synthesis and biological evaluation of novel 2-aminobenzamides containing dithiocarbamate moiety as histone deacetylase inhibitors and potent antitumor agents[J]. Eur J Med Chem, 2018, 143: 320-333. |
| [32] |
Altintop MD, Sever B, Ciftci GA, et al. Synthesis and evaluation of new benzodioxole-based dithiocarbamate derivatives as potential anticancer agents and hCA-I and hCA-II inhibitors[J]. Eur J Med Chem, 2017, 125: 190-196. |
| [33] |
Qian Y, Ma GY, Yang Y, et al. Synthesis, molecular modeling and biological evaluation of dithiocarbamates as novel antitubulin agents[J]. Bioorg Med Chem, 2010, 18: 4310-4316. |
| [34] |
Fu DJ, Zhang SY, Liu YC, et al. Design, synthesis and antiproliferative activity studies of novel dithiocarbamate-chalcone derivates[J]. Bioorg Med Chem Lett, 2016, 26: 3918-3922. |
| [35] |
Wang XJ, Xu HW, Guo LL, et al. Synthesis and in vitro antitumor activity of new butenolide-containing dithiocarbamates[J]. Bioorg Med Chem Lett, 2011, 21: 3074-3077. |
| [36] |
Huang W, Ding Y, Miao Y, et al. Synthesis and antitumor activity of novel dithiocarbamate substituted chromones[J]. Eur J Med Chem, 2009, 44: 3687-3696. |
| [37] |
Duan YC, Ma YC, Zhang E, et al. Design and synthesis of novel 1, 2, 3-triazole-dithiocarbamate hybrids as potential anticancer agents[J]. Eur J Med Chem, 2013, 62: 11-19. |
| [38] |
Duan YC, Zheng YC, Li XC, et al. Design and synthesis of novel 1, 2, 3-triazole-dithiocarbamate hybrids as potential anticancer agents[J]. Eur J Med Chem, 2013, 64: 99-110. |
| [39] |
Ye XW, Zheng YC, Duan YC, et al. Synthesis and biological evaluation of coumarin-1, 2, 3-triazole-dithiocarbamate hybrids as potent LSD1 inhibitors[J]. Med Chem Commun, 2014, 5: 650-654. |
| [40] |
Cao SL, Feng YP, Jiang YY, et al. Synthesis and in vitro antitumor activity of 4(3H)-quinazolinone derivatives with dithiocarbamate side chains[J]. Bioorg Med Chem Lett, 2005, 15: 1915-1917. |
| [41] |
Cao SL, Yao W, Zhu L, et al. Synthesis and cytotoxic activity of N-((2-methyl-4(3H)-quinazolinon-6-yl)methyl) dithiocarbamates[J]. Eur J Med Chem, 2010, 45: 3850-3857. |
| [42] |
Cao SL, Han Y, Yuan CZ, et al. Synthesis and antiproliferative activity of 4-substituted-piperazine-1-carbodithioate erivatives of 2, 4-diaminoquinazoline[J]. Eur J Med Chem, 2013, 64: 401-409. |
| [43] |
Fu DJ, Zhang L, Song J, et al. Design and synthesis of formononetin-dithiocarbamate hybrids that inhibit growth and migration of PC-3 cells via MAPK/Wnt signaling pathways[J]. Eur J Med Chem, 2017, 127: 87-99. |
| [44] |
Yu JY, Li XQ, Wei MX. Synthesis and biological activities of artemisinin-piperazine-dithiocarbamate derivatives[J]. Eur J Med Chem, 2019, 169: 21-28. |
| [45] |
Lane CA, Hardy J, Schott JM. Alzheimer's disease[J]. Eur J Neur, 2018, 25: 59-70. |
| [46] |
Yang WC, Sun Q, Yu NX, et al. Design of acetylcholinesterase inhibitor for Alzheimer's disease therapy:from multi-bindingsite inhibitors to multi-target directed ligands[J]. Acta Pharm Sin (药学学报), 2012, 47: 313-321. |
| [47] |
Ferreira-Vieira TH, Guimaraes IM, Silva FR, et al. Alzheimer's disease:targeting the cholinergic system[J]. Curr Neuropharmacol, 2016, 14: 101-115. |
| [48] |
Altintop MD, Gurkan-Alp AS, Ozkay Y, et al. Synthesias and biological evaluation of a series of dithiocarbamates as new cholinesterase inhibitors[J]. Arch Pharm, 2013, 346: 571-576. |
| [49] |
Altintop MD, Ozdemir A, Kaplancikli ZA, et al. Synthesis and biological evaluation of some pyrazoline derivatives bearing a dithiocarbamate moiety as new cholinesterase inhibitors[J]. Arch Pharm, 2013, 346: 189-199. |
| [50] | |
| [51] |
Jiang N, Huang QC, Liu J, et al. Design, synthesis and biological evaluation of new coumarin-dithiocarbamate hybrids as multifunctional agents for the treatment of Alzheimer's disease[J]. Eur J Med Chem, 2018, 146: 287-298. |
| [52] |
He Q, Liu J, Lan JS, et al. Coumarin-dithiocarbamate hybrids as novel multitarget AChE and MAO-B inhibitors against Alzheimer's disease:design, synthesis and biological evaluation[J]. Bioorg Chem, 2018, 81: 512-528. |
| [53] |
Jiang N, Ding JL, Liu J, et al. Novel chromanone-dithiocarbamate hybrids as multifunctional AChE inhibitors with β-amyloid anti-aggregation properties for the treatment of Alzheimer's disease[J]. Bioorg Chem, 2019, 89: 1-12. |
| [54] |
Fu J, Bao FQ, Gu M, et al. Design, synthesis and evaluation of quinolinone derivatives containing dithiocarbamate moiety as multifunctional AChE inhibitors for the treatment of Alzheimer's disease[J]. J Enzym Inhib Med Chem, 2020, 35: 118-128. |
| [55] |
Liddell JR, Lehtonen S, Duncan C, et al. Pyrrolidine dithiocarbamate activates the Nrf2 pathway in astrocytes[J]. J Neuroinflamm, 2016, 49: 2-14. |