 2020, Vol. 55
2020, Vol. 55

·新药发现与研究实例简析·
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
艾拉戈克是作用机制明确的药物, 靶标是促性腺激素释放激素(GnRH)受体。由于人体的下丘脑-垂体-性腺轴系统的生理功能已完全清晰, 其病理过程和药物治疗原理因而也是明确的。但由于受体的三维结构尚未解析, 用药物化学方法研制小分子抑制剂比肽类药物难度大得多。研制本品没有用特殊的方法和技术, 但围绕药效活性、药代稳定性和口服的物化性质等方面对先导物的优化可谓“地毯式”的探索与修饰, 对每一个结构片段可谓做到精雕细刻地修饰, 这可从发表的大量药物化学论文得到佐证。从2002年发表在J Med Chem第一篇论文算起, 到2018年批准上市, 研制时程估计经20年, 彰显了这一首创的治疗子宫内膜异位症的口服药物的风险与艰辛。)
(编者按)
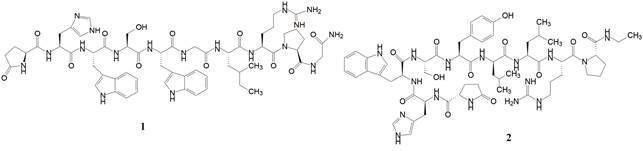
本项目是通过干预下丘脑-垂体-性腺轴系统治疗与性器官相关的疾病, 临床治疗子宫内膜异位症。下丘脑-垂体-性腺轴是控制人体性激素分泌的一个分支。下丘脑分泌促性腺激素释放激素[gonadotropin-releasing hormone (GnRH)], 又称黄体生成激素释放激素(LH-RH), GnRH在脑垂体刺激GnRH受体, 促进垂体分泌促黄体生成素(LH)、促卵泡刺激素(FSH)和泌乳素(PRL)等。
GnRH是个线型十肽化合物(1), 子宫内膜异位和前列腺癌等一些性器官疾病可通过抑制GnRH受体加以治疗和缓解。临床应用的GnRH受体拮抗剂是肽类药物, 例如亮丙瑞林(2, leuprorelin)治疗子宫内膜异位症、前列腺癌和中枢性性早熟症等。
然而肽类拮抗剂疗法具有反跳性(flare effect), 即用药后最初产生激活的不良反应, 然后才出现抑制活性, 而且需注射治疗。人们期望用小分子拮抗剂克服反跳作用和只能注射用药等缺点。
2 活性评价体外评价受试物与受体的结合性能的方法是在96孔板稳定表达人GnRH受体的HEK293细胞, 与125I标记的合成GnRH类似物[(r125I-Tyr5, DLeu6, NMeLeu7, Pro9-Net) GnRH]以及不同浓度的受试物共同进行温孵, 测定受试物竞争性结合受体的作用, 计算结合常数(Ki), Ki值越小结合作用越强。
评价受试物对受体的抑制性实验, 是在96孔板上稳定表达人GnRH受体的RBL-1细胞, 加入胎牛血清、营养液和3H标记的肌醇, 加入不同浓度受试物温孵, 洗涤并溶解细胞后, 液闪测定放射性计数, 计算受试物抑制50%磷酸肌醇生成的浓度(IC50)。
|
Neurocrine公司(后被艾伯维收购)的先导化合物是3和4, 首轮变换3个位置的取代基: 1位加入碱性氮原子以提供氢键结合, 2位苯环上的烷氧基的变换和4位连接疏水性基团以优化疏水性结合。合成的代表性化合物列于表 1。

|
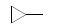
分析表 1化合物的共同特征是, R2的芳环和R3的碱性基团都是经-CH2-与母核连接, 这样就避免了共轭体系的形成, 不会降低氮原子的碱性, 也避免了单键连接两个芳环而刚性过强。分析构效关系如下: ① R2的苯环上取代基变换对活性影响不大, 例如化合物8~10的活性未见提升, 但2-氟代的11 (Ki = 0.5 μmol·L-1)活性强于7, 可能是氟原子的电性和体积最适配于结合。再添加1个氟原子, 二氟苄基化合物12~15的活性反而降低了。② R3的长链是必要的。化合物5是二甲氨基, 没有活性(Ki > 50 μmol·L-1), 若1个甲基用苄基取代, 化合物6只有边缘活性(Ki = 42 μmol·L-1), 但甲基若被2-(2-吡啶基)乙基置换, 活性显著提高, 7的Ki = 2.6 μmol·L-1, 其他有该侧链的化合物的活性都较强, 提示R3处有一个疏水腔, 并且氢键的形成有利于结合。③基于R2位2-氟苯基对活性的正贡献, 再检讨R3的侧链的构效关系, 化合物17没有吡啶基, 活性虽不如11, 但显著强于6, 反证了2-氟苯基为优化片段。④ R1的甲基变成亲脂性强的异丁基, 活性普遍增强, 例如18强于10, 19~22强于相应的11、13~15。⑤ R3的2-吡啶乙基换成3-或4-吡啶乙基的活性(23, 24)显著减弱, 提示2-吡啶氮原子位置对结合的重要性。
3.2 R1和羧酸酯的变换下一步研究的构效关系是, 固定2-吡啶乙胺基和2-氟代苄基不变, 探索2位苯环上其他烷氧基和6位羧酸的酯基对活性的影响, 合成的代表性化合物列于表 2。结果表明, 由乙酯11 (Ki = 500 nmol·L-1)变为异丙酯(26)活性基本未变(Ki = 400 nmol·L-1), 而换成环戊酯(27, Ki = 260 nmol·L-1)或3-戊酯(39, Ki = 180 nmol·L-1)活性提高2倍, 但再大的酯基如二环丙甲基(28, Ki = 2 500 nmol·L-1)或四氢呋喃酯(29, Ki = 2 100 nmol·L-1)活性减弱, 提示酯基宜有适宜的尺寸, 过大或过小不利。总结这些位置的变换的综合效果, 化合物31活性最强, Ki = 25 nmol·L-1, 类似结构的32和33也有高活性。应当指出, 化合物对人与大鼠的GnRH活性有显著不同, 例如30~33对大鼠活性很弱, 提示GnRH具有种属差异性(Zhu YF, Struthers RS, Connors PJ, et al. Initial structure-activity relationships of a novel series of pyrrolo[1, 2-a]pyramid-7-ones as GnRH receptor antagonists. Bioorg Med Chem Lett, 2002, 12: 399-402)。
| Table 1 Binding affinities of pyrrolo[1, 2-a]pyrimidones on the human GnRH receptor |
| Table 2 Binding affinities of compounds with varied R1 and carboxylic ester |
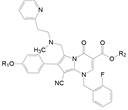
在结构变换中另一个路经是考证杂环母核上的氰基的必要性, 探索1-位碱性侧链、2位苯基上的取代基、4-位N-取代基的变换对活性的影响(6位羧基均固定为乙酯), 结果发现2位苯环上含有4-丁酰氨基化合物34具有高活性, Ki = 1.2 nmol·L-1 (过程从略)。进而以34为新的起点将6位的羧酸酯变换为酰胺以提高化合物的稳定性, 合成的有代表性化合物列于表 3。
| Table 3 Binding affinities of compounds with amide |
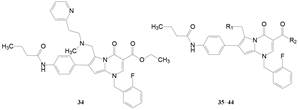







结果表明, 氰基对活性的贡献不大。酰戊胺35的活性与酰环戊胺36相比, 后者活性增强一倍, 扩环成环己胺38的活性降低两倍。开链的正戊胺39活性显著提高, Ki = 3.3 nmol·L-1, 丁胺(37)的活性略弱, 含甲氧基时(40)降低很多, Ki = 630 nmol·L-1。苯丙胺(41)活性强于34, 并且R1为甲基苄胺(43)的活性降低, 说明2-吡啶乙基胺侧链在酰胺系列中仍是重要的(Zhu YF, Wilcoxen K, Saunders J, et al. A novel synthesis of 2-arylpyrrolo[1, 2-a]pyrimid-7-ones and their structure-activity relationships as potent GnRH receptor antagonists. Bioorg Med Chem Lett, 2002, 12: 403-406)。
然而, 吡咯并嘧啶酮母核没有拉电子的氰基, 使得化合物的代谢稳定性降低, 半衰期很短, 仍需继续优化。
5 咪唑并嘧啶酮母核 5.1 咪唑并嘧啶酮环上连接基的优化用咪唑并嘧啶酮代替吡咯并嘧啶酮母核(将原来的氰基氮原子融到并环中), 可增加极性, 以提高化合物的代谢稳定性。将前述各个位置优化的基团连接在新的母核上, 合成的化合物列于表 4。
| Table 4 Structure-activity relationships for the 2-arylimidazolo[1, 2-a]pyrimid-5-ones |
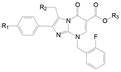
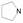
解析表 4的构效关系提示: ①化合物45与46的活性差异再次表明R2含有2-吡啶的侧链优于苄胺侧链; 比较47与45, R1的丁酰胺显著优于甲氧基, 活性提高11倍, 提示是疏水性和(或)氢键给体起作用的缘故。48强于47大约6倍, 是2-吡啶乙基侧链的贡献。这些说明, 前面述及的优势片段连接在新的母核仍然具有有效性。②乙基酯(C2酯)换成2-乙基丙酯(C5酯)活性增高, 例如成对的化合物49强于48, 57强于47, 说明酯基片段的大体积和疏水性有利于活性。③ R2的2吡啶乙基被3-吡啶甲基(51)或2-呋喃甲基(52)或甲氧乙基(53)替换, 活性都有所减弱, 而被氰甲基(54)、四氢异喹啉(55)和四氢吡咯(56)替换, 则失去活性(Wilcoxen KM, Zhu YF, Connors PJ Jr, et al. Synthesis and initial structure-activity relationships of a novel series of imidazolo[1, 2-a]pyrimid-5-ones as potent GnRH receptor antagonists. Biooeg Med Chem Lett, 2002, 12: 2179-2183)。
5.2 酯基的消除性变换化合物49是高活性化合物, Ki = 7.5 nmol·L-1, 3-戊基酯的作用推测是亲脂性和氢键接受体, 但它的化学或代谢稳定性差, 这一轮的优化是用稳定的基团替换酯基, 即用带有取代基的苯基取代原酯基的功能, 合成的化合物列于表 5。
| Table 5 tructure-activity relationships of compounds without ester groups |
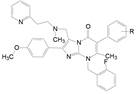
甲氧苯基满足上述疏水和氢键接受体的条件, 化合物58的活性(Ki = 4.6 nmol·L-1)超过了49, 提示设计的合理性, 而且将甲氧基移至2-或4-位(59, 60), 活性减弱, 说明氧原子位置的重要性, 2位比其他位置有利于结合。苯环上含有两个或3个甲氧基的化合物(62, 63)活性都降低10倍以上, 3, 4-亚甲二氧化合物61的活性仅次于58, 卤素替换甲氧基的化合物活性都较低, 是因为卤素电负性高, 难以作为氢键的接受体。
进而固定R为亚甲二氧基, 变换2-吡啶乙胺基侧链为无吡啶环的疏水链, 活性都显著下降, 提示羧酸酯换成取代苯的化合物仍以2-吡啶乙胺基为优化片段(结构与数据从略) (Gross TD, Zhu YF, Saunders J, et al. Design, synthesis and structure-activity relationships of novel imidazolo[1, 2-a]pyrimid-5-ones as potent GnRH receptor antagonists. Bioorg Med Chem Lett, 2002, 12: 2185-2187)。
6 简化稠合环为单环 6.1 咪唑并嘧啶酮变换为尿嘧啶环为了简化母核结构, 将稠合环变为单环, 去除2位苯环和连接它的碳原子, 使其成为尿嘧啶结构, 从而省略了两个环, 以有利于药代和物化性质, 合成的代表性化合物列于表 6。结果表明, 母核缩减后, 化合物68~71的侧链结构与活性的关系与原来稠合环的规律基本相同, 仍以2-吡啶乙胺基(68)活性最高, 碳链缩短(69)或吡啶环换成苯环(70)或咪唑环(71)都使活性降低。而中间氮原子成环的化合物(72~74)活性更显著降低。化合物68是活性强的化合物。此外, 为了考察尿嘧啶6位甲基的活性贡献, 合成的无甲基物75和6-乙基物76的活性分别降低50和65倍, 说明6-甲基是非常必要的药效基团。
| Table 6 Structure and activity of compounds with uracil core |
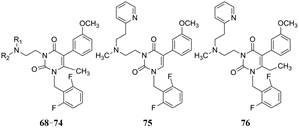
为了考察化合物68的5-苯基环上3′-甲氧基的重要性, 去除甲氧基的化合物77活性显著下降(表 7)。而3, 4-亚甲二氧基(78)和3, 4-亚乙二氧基(79)取代的活性与68相似, 甲氧基移至4位(83)活性降低6倍, 提示间位的必要性, 推测是履行氢键接受体的功能。3-OH (80)或3-OCF3 (81)等极性基团没有活性, 4-苯氧基(84)与68相近, 可能是苯基π电荷提供了用于结合的部分电荷。
| Table 7 Activity of compounds with varied substituted 5-phenyl groups |
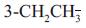
高活性的68是里程碑化合物, 然而小鼠和人肝微粒体温孵提示它的代谢稳定性差, 灌胃小鼠表明75%的药量被肝脏代谢, 血液中药物的消除半衰期0.4 h, 口服生物利用度F=1.4%, 这些表明脂溶性较强的68的药代尚需优化(Zhu YF, Gross TD, Guo ZQ, et al. Identification of 1-arylmethyl-3-(2-aminoethyl)-5-aryluracil as novel gonadotropin-releasing hormone receptor antagonists. J Med Chem, 2003, 46: 2023-2026)。
有趣的是2-F化合物(85)活性显著高于3-F (86), 因而合成了2-F, 3-OCH3 (87)活性是68的10倍, 从而开辟了5位苯环上双取代的途径。在这里体现了基团的加和效应。
6.3 苯环取代基和含氮侧链的再变换由于5位苯基的2-F对活性的有益贡献以及N3侧链具有可变换空间, 确定继续优化这两个位置, 即变换N3链的同时, 再探索2-F代苯环的影响, 合成有代表性的化合物列于表 8。构效关系表明: ①化合物88是将68的吡啶乙基缩短为苯甲基, 并在N3链的β位连接甲基(链上碳数不变), 因而88成为手性分子, 证明对映体之间的活性基本相同, 但R-88活性低于68, 而R-89 (吡啶甲基)活性则强于68一倍, 提示将链上的一个甲基迁移成支链的R-构型对结合的重要性。② 89的吡啶甲基加长为吡啶乙基(90)的活性降低, 例如R-优映体的活性降低5倍。③将88的5-(3-甲氧苯基)上引入2-F基团, 化合物91的活性显著提高, 其S异构体活性接近R-89, 说明氟原子的引入提高了整体分子与受体的结合能力, 而且构型不同。④ R-92则由于氟原子和吡啶的协同(或加和)作用活性达到Ki = 1.1 nmol·L-1。其实, 苯环上2-F-3-OCH3的取代, 使得含氮侧链末端换成不同的杂环或支链烷基或环烷基的化合物(93~97), R或S构型的活性都很高。进一步变β-甲基为α-甲基的化合物虽然仍以R构型为优映体, 但活性有所降低(结构和数据省略) (Guo ZQ, Zhu YF, Gross TD, et al. Synthesis and structure-activity relationships of 1-arylmethyl-5-aryl-6-methyluracils as potent gonadotropin-releasing hormone receptor antagonists. J Med Chem, 2004, 47, 1259-1271)。
| Table 8 Binding affinity of compounds with varied N3- and C5-substituents |
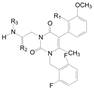


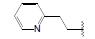
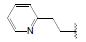
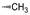
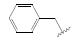
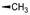
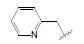


化合物R-92以及类似的高活性化合物(例如环戊基等)存在的主要问题是代谢不稳定性, 与人肝微粒体温孵被迅速氧化脱除N-烷基成伯胺, 代谢物因亲脂性降低而活性减弱。为避免该代谢失活作用, 将连接在氮上的基团移至侧链的β位, 使氮原子成为伯胺, 以避免氧化代谢, 且保持侧链的亲脂性, 设计合成的化合物列于表 9。表中成对的对映体活性表明, 烷基或环烷基取代的R构型强于相应的S构型, 例如R-环己基(R-100)和R-叔丁基(R-101)的活性高达Ki = 6 nmol·L-1。然而苄基取代的化合物103的优映体是S构型。苄基增加一个碳原子成苯乙基(S-104)的活性显著下降, 提示R构型的基团进入的结合腔容积有限度。在所有的化合物中R-苯基化合物R-105活性最强(Tucci FC, Zhu YF, Guo ZQ, et al. 3-(2-Aminoalkyl)-1-(2, 6-difluorobenzyl)-5-(2-fluoro-3-methoxyphenyl)-6-methyluracils as orally bioavailable antagonists of the human gonadotropin releasing hormone receptor. J Med Chem, 2004, 47: 3483-3486)。
| Table 9 Binding affinity of compounds with varied β-substituted groups |
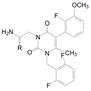
虽然避免了氧化脱烷基的不利因素, 如前述及, 伯胺一般比相应的仲胺活性弱, 为了进一步提高活性, 将伯胺再作烷基化, 合成的代表性化合物列于表 10。分析构效关系如下: ①化合物S-109和S-115分别是S-103的N-单甲基和二甲基化合物, 活性都有所提高。而高活性的伯胺R-101 (Ki = 6.4 nmol·L-1)经N-甲基化, R-108 (异丁基而非叔丁基)的活性提高5倍。有趣的是苄基化合物的S构型是优映体, 而异丁基的优映体是R构型。②环戊基化合物113的两个对映体活性基本没有区别, 都非常高, 提示基团的变化对于对映体之间活性的影响是不一样的。③苯基化合物的氨基被单或双甲基化活性略有减弱, 其中R-116的活性较高, 但仍没有超过相应的伯胺化合物R-105 (Tucci FC, Zhu YF, Struthers RS, et al. 3-[(2R)-Amino-2-phenylethyl]-1-(2, 6-difluorobenzyl)-5-(2-fluoro-3-methoxyphenyl)-6-methylpyrimidin-2, 4-dione (NBI 42902) as a potent and orally active antagonist of the human gonadotropin-releasing hormone receptor. Design, synthesis, and in vitro and in vivo characterization. J Med Chem, 2005, 48: 1169-1178)。
| Table 10 Binding affinity of compounds with varied β-substituted and amino groups |
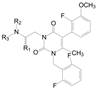
前面优化中固定了N1位是2, 6-二氟代苄基, C5位是2′-F-3′-OCH3, 活性最高的化合物是N3-R-β-氨基苯乙基化合物(R-105), 其实, N1和C5的取代基没有进行系统的优化, 为此固定N3为R-β-氨基苯乙基, 而且为了合成的方便, 模板化合物为R-118, 即母核上C-6没有甲基的R-105, R-118的活性逊于R-105。这样得到的构效关系可以直接套用在含6-甲基的系列中, 这是药物化学常用的方法。合成代表性化合物列于表 11。
| Table 11 Binding affinity of compounds with varied substituted N1- and C5-groups |
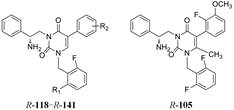
表 11的构效关系揭示如下: ①手性分子的R仍比S构型的活性高, 例如118和124的R构型是优映体。② N1连接的苄基环上6位连接强拉电子基团, 活性强于2, 6-二氟化合物, 例如三氟甲基、甲磺酰基和氯代为强活性的取代基, 而推电子基团如甲硫基(R-125)和极性基团(R-128~R-130)活性则降低。③ C5相连的苯基上仍以2-F-3-OCH3取代基为优选基团。④活性最强的化合物是R-124, 从而合成了对应的6-甲基化合物(R-142)。
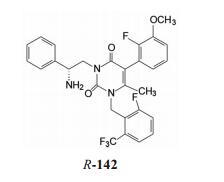
|
化合物R-142的Ki值为0.56 nmol·L-1, 与相应的二氟化合物R-105对受体的结合活性相同(Ki = 0.56 nmol·L-1), 不过对受体的功能性抑制作用表明, R-142与R-105的IC50分别是0.50 nmol·L-1和2.8 nmol·L-1, 相差6倍。此外, 还用结合动力学方法比较了这两个化合物对hGnR H受体结合成复合物的离解速率(慢离解速率表明作用的持续性长), 表明R-142的结合半衰期t1/2 > 43 h, 长于R-105的t1/2 = 4.3 h的10倍, 提示这个C6-CH3改变了结合后受体的构象, 显著降低了复合物的离解速率。
虽然R-142是hGnRH受体强效的拮抗剂, 但发现对CYP3A4酶呈现抑制作用, IC50 = 0.23 µmol·L-1, 这种药效和药代的“冲突”需要进一步变换结构来化解。其中一个方法是增加分子的极性, 例如引入极性基团或电荷(形成两性离子)以降低与CYP的结合。研制者最初在C5-的苯环加入了极性基团如羟甲基、胺基、羧烷基等替换2-F-3-OCH3基团, 虽然降低了对CYP3A4的抑制作用, 但也减弱了抑制hGnRH受体的活性, 总效应得不偿失。但指明了极性基团可降低抑制CYP3A4作用的出路(数据省略) (Chen C, Chen YS, Pontillo J. Potent and orally bioavailable zwitterion GnRH antagonists with low CYP3A4 inhibitory activity. Bioorg Med Chem Lett, 2008, 18: 3301-3305)。
另一个途径是在氨基上做极性链的取代。由于前面的构效关系揭示伯胺的单取代成仲胺的活性变化不大, 因而合成了N-羧烷基化合物, 结构和活性列于表 12。这些化合物测定了与hGnRH受体的结合作用和拮抗活性, 以及对CYP3A4的抑制作用。变换N-烷酸的链长就是找到对hGnRH受体(靶向)和CYP3A4 (脱靶)的最佳平衡点, 以最大化于药理活性。结果表明, R-146对受体的抑制作用虽然比R-142减弱2倍(IC50 = 1.5 nmol·L-1), 但抑制CYP3A4的作用降低了近250倍(IC50 = 56 μmol·L-1), 围绕R-146合成的化合物如将羧基甲酯化(R-143)后对CYP3A4的抑制活性仍很强, 是因为脂溶性高的缘故; N-戊酸化合物(R-147)或N-再甲基化成叔胺(R-148)导致抑制受体作用降低, 此外, 变换R-146的其他基团, 如三氟甲基换成氟或氯原子都使活性降低(数据省略)。C6-去甲基化合物R-149的抑制受体活性也显著降低, 因此化合物R-146是优化最好的化合物。
| Table 12 Structure, activity and CYP3A4 inhibition of R-N-carboxyalkyl compounds |
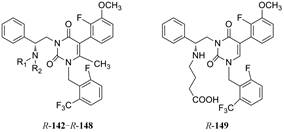
为了慎重选取候选化合物, 对高活性的化合物R-142、R-146和R-148进行了体内外活性、药代和物化性质的系统评价, 表 13列出了体外稳定性、过膜性和分布系数。由于连接丁酸链的R-146的清除率降低, 提示对人肝微粒体的稳定性提高, 进一步N-甲基化的R-148稳定性则下降。丁酸基团的引入使得向Caco-2细胞渗透的能力降低, 是因为两性离子不利于穿越脂质性细胞膜。比较化合物从基侧到顶端方向与顶端到基侧方向的外排比值(efflux ratio)表明, R-146 (不利)的外排作用强于R-148。
| Table 13 Metabolic stability in human liver microsomes and permeability in Caco-2 cells. Digoxin was used in the Caco-2 assay as a reference, which had an average Papp of 10 nm·s-1 and (b to a)/(a to b) ratio of ∼20 |
进而用大鼠、比格犬和恒河猴比较了3个高活性化合物的药代动力学性质, 列于表 14。结果表明, R-146与R-148在不同的实验动物的药代动力学性质差别较小, R-146只是在比格犬的口服生物利用度显著高于R-148, 药效学实验表明R-146可逆性抑制猴血浆中促黄体生成素(LH)水平。此外R-146对CYP1A2、CYP2D6、CYP2C9和CYP2C19等药物代谢酶没有显示抑制作用。
| Table 14 Comparison of PK parameters of selected compounds on animals |
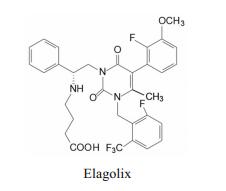
综合化合物的活性和药代性质, 确定了化合物R-146为候选化合物, 定名为艾拉戈克(elagolix), 经III期临床研究, 表明可治疗子宫内膜异位症, 为作用于GnRH受体的口服药物。在2018年经美国FDA批准上市(Chen C, Wu DP, Guo ZQ, et al. Discovery of sodium R-(+)-4-{2-[5-(2-fluoro-3-methoxyphenyl)-3-(2-fluoro-6-[trifluoromethyl]-benzyl)-4-methyl-2, 6-dioxo-3, 6-dihydro-2H-pyrimidin-1-yl]-1-phenylethylamino}butyrate (Elagolix), a potent and orally available nonpeptide antagonist of the human gonadotropin-releasing hormone receptor. J Med Chem, 2008, 51: 7478-7485)。
|
纵观艾拉戈克的历程, 涉及的头绪很多, 先导化合物的各个部分经过全盘的变换和优化使分子面目皆非, 有些部位进行了反复验证, 在药效、药代和物化性质等获得最佳的容许度。图 1简要地总结了研发过程的重要节点。

|
Figure 1 Important points in R & D process of elagolix |