 2020, Vol. 55
2020, Vol. 55



2. 中国药科大学药学院, 江苏 南京 210009
2. School of Pharmacy, China Pharmaceutical University, Nanjing 210009, China
在任何时期, 新药创制都关乎人们的生命健康与生活质量。新药创制是一项极其复杂的系统工程, 同时涵盖基础研究、生产开发、市场销售等各个环节, 需要巨大的经济投入并伴随着极高的研发风险。在新药的创制过程中, 基于药物靶标的新颖性特征可将创新药物分为首创性药物(first-in-class)和跟随性药物(me too, me better), 二者对新药创制的过程都具有巨大的推动作用。其中, 首创性药物需要作用于全新的候选靶标, 随后针对其靶标特性合理设计药物分子, 以实现对某种疾病的治疗效果, 在研发的过程中依靠生物学驱动。重要的是, 首创性药物的靶标再确证过程贯穿药物研发始终, 从前期的实验室研究阶段到药物上市后的应用, 都在反复验证全新靶标的安全性、有效性以及和疾病间的因果性。因此, 首创性药物的研发难度高、周期长、投入多、风险大, 但一旦成功往往也伴随着丰厚的回报。跟随性药物主要针对已有药物的缺陷, 有目的地进行药物的改良, 亦或为了突破专利以打破市场的垄断。由于跟随性药物的生物学功能相对明晰, 研发过程往往基于化学结构的修饰或药物骨架的突破, 主要依靠化学驱动。研发跟随性药物需要找到与同类药物的作用差异, 解决不同的临床需求或治疗效果优于首创性药物, 由于需要保证开发速度, 及早占领市场, 因此面临很强的竞争性[1]。
在首创性药物的研发过程中, 全新作用靶标的发现至关重要。目前, 化学生物学的研究可以为全新靶标的发现提供重要的早期研究证据, 其研究过程聚焦于发现潜在的功能蛋白, 并使用化学手段获得可以调控其功能的工具分子。合理使用此类工具分子深入研究功能蛋白的生物学机制, 可将这些功能蛋白与相应的疾病关联。经过反复研究后具有成药性的功能蛋白成为潜在的药物靶标, 前期研究中的工具分子成为苗头化合物。为了实现对潜在药物靶标的高效调控, 苗头化合物需要经过药物化学工作的不断优化, 涵盖其活性优化、理化性质优化、成药性优化等环节, 最终获得高活性、高特异性并具有良好成药性的先导化合物分子, 进而开展后续的临床前和临床研究。由此可见, 首创性药物的研发以功能蛋白和工具分子的发现为起始, 经历生物学机制的反复探索, 逐步过渡至潜在的药物靶标与苗头化合物, 随后还需经过漫长的分子优化过程与靶标再确证, 最终才可能进入临床研究。首创性药物往往需要漫长的研发周期, 例如2016年全球第一个蛋白-蛋白相互作用抑制剂维奈克拉(Venetoclax)的上市具有教科书般的重要意义, 其研发过程曲折多变, 从发现苗头到成功上市历经20余年, 综合运用了多种药物研究技术, 打破了传统的类药五规则, 在药物靶标、药物分子结构、蛋白-蛋白相互作用抑制剂的设计方法等方面引入全新理念, 开启了后续蛋白-蛋白相互作用抑制剂的研究热潮。但首创性药物漫长的研发过程往往伴随着极高的失败风险, 例如基于包括β-淀粉样蛋白、5HT6和PPARγ等“全新”靶标在内的抗老年痴呆(AD)药物成为了新药创制的重灾区, 众多分子在临床试验中夭折, 至今尚无成功的研究案例。同时, 由于首创性药物需要通过新靶标、新机制而起效, 具有潜在的机制杂泛性和通路上下游的机制未知性, 更需要考察其不良反应所带来的风险。历史上著名的降胆固醇药物CETP抑制剂先后有4个研究中新药进入Ⅲ期临床, 开展了上万例患者的临床试验, 但由于引起了少数心脏不良反应及猝死事件, CETP最终不能成为药物靶标以失败而告终, 以辉瑞公司为代表的制药巨头投入了超过10亿美元的研发资金, 耗时超过15年。
确证药物靶标与对应疾病之间的因果性关系对首创性药物极其关键, 这意味着对特定疾病的治疗效果是否真实地基于药物靶标而实现, 而非通过其他未知途径起效。但是, 因果性的验证非常困难。在目前靶向药物的研究中, 为了更快推进药物的研发进程, 往往只关注与作用靶标相对应的生物标志物的降低或升高, 以及药物是否取得了预想的生物学效应。然而, 如果不阐明药物靶标与治疗效果之间的因果性, 即意味着有潜在的“炸弹”尚未被发现。新药研发历史上著名的“反应停”事件、罗非昔布的撤回事件等都证明成功的药物需要长时间的反复验证。首创性药物需要全新的作用靶标, 更需要深入研究全新靶标与治疗效果之间的未知性, 阐明其因果性, 是一项极具挑战性的工作。
2019年, 美国FDA和CDER共批准上市了48个新药, 尽管少于2018年的59个, 但仍可以排在近25年来批准新药数量的第3位。2019年获批的新药中有28个获得优先审批资格, 旨在提供对现有重大疾病的治疗选择; 通过突破疗法和加速审批的上市新药分别为13个和9个; 孤儿药获批21个, 保持了迅猛增长的态势, 表明创新药物的研发不仅聚焦于解决重大疾病难题, 也开始更多地涉足到各类孤儿病的治疗领域。鉴于孤儿病的特征, 其药物研发往往需要首创性药物。随着生物技术的不断发展, 各类大分子的生物药研究势头强劲, 但小分子药物在目前的新药开发中仍是研究的重点。在2019年的48个获批新药中, 38个是新分子实体(NMEs), 10个是生物药物(部分获批的小分子药物总结于表 1)。本文选取了3个具有代表的首创性小分子药物进行介绍, 通过研发背景、药物设计思路及治疗应用等方面浅析其研发过程, 为更多首创性药物的研发提供参考。
| Table 1 Small molecule drugs approved by FDA in 2019. CABP: Community-acquired bacterial pneumonia; SCD: Sudden cardiac death; IBS: Irritable bowel syndrome |
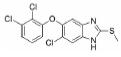
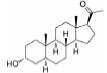
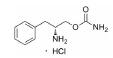
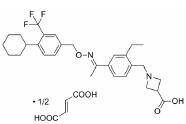
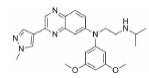
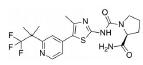
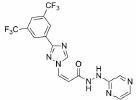
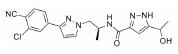
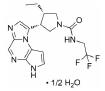
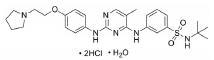
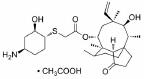
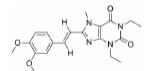
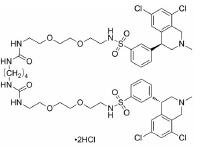
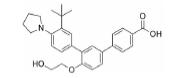
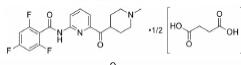
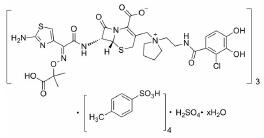
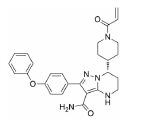
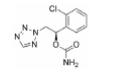
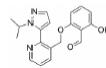
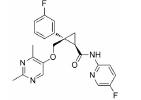
根据美国国家癌症研究所(National Cancer Institute, NCI)的统计数据, 在美国多发性骨髓瘤患者有13万人, 并以每年新增3.2万的速度持续增加, 2019年美国死于多发性骨髓瘤的患者达到1.3万人[2]。针对这一棘手的疾病, 目前的治疗策略主要包括多种蛋白酶体抑制剂药物和免疫调节剂, 患者在接受治疗后仍会复发, 也有部分患者对现有的治疗策略无效, 针对此情况尚无有效的药物治疗方案。
在细胞的正常功能中, 核转运蛋白负责将细胞核内的各类信号因子转运出细胞核, 使其被识别并发挥生物学功能[3]。核转运蛋白的活性主要依赖各类运输蛋白(transport proteins, exportins)。其中, 核输出蛋白XPO1 (nuclear export protein exportin1, 又名CRM1)介导的输出蛋白约220种, 可以特异性地实现多种肿瘤抑制因子(tumor suppressor, TSP)和生长调控蛋白(growth regulatory proteins, GRP)的核运输过程, 其中包括p53、p21、p73、Rb1、APC、Bcl-Abl、FOXO和STAT3等肿瘤相关蛋白[4, 5]。在正常的生理条件下, 通过核输出蛋白转运此类蛋白可以在DNA损伤或其他致癌情况下避免致癌基因的过表达, 维持生理稳态。在肿瘤细胞中, 将此类蛋白转运出细胞核会抑制肿瘤抑制因子的活性, 促进肿瘤的发生。在多种血液癌和实体瘤中, 都观察到了XPO1的过表达现象[6]。其作用机制如图 1所示, XPO1结合Cargo蛋白(被转运的蛋白总称, 包括肿瘤抑制因子、抗凋亡蛋白等), 通过形成Ran-GTP中间态将目标蛋白转运出细胞膜, 随后XPO1解离后返回细胞核, 完成一个循环周期。选择性抑制XPO1会使细胞核内的TSPs和GRPs积聚, 可以抑制肿瘤的发生与发展, 提供了一种全新的抗肿瘤药物设计策略与机制。

|
Figure 1 Biological mechanism of XPO1 for transportation of cargo from nucleus to cytoplasm. NPC: Nuclear pore complex |
鉴于XPO1重要的生物学功能, 其抑制剂的研发也受到了广泛的关注, 目前已有多个抑制剂正在进行临床研究。2019年7月, 塞利尼索获批成为全球首个口服选择性核输出抑制剂(selective inhibitor of nuclear export, SINE), 通过直接靶向于核输出蛋白XPO1而起效。塞利尼索获得了加速审批和孤儿药资格, 首次证明了靶向XPO1在复发的多发性骨髓瘤中的应用, 属于首创性药物。XPO1作为靶标的生物学功能发现于1997年, 距离其首个抑制剂成功上市长达22年, 首创性药物的研发难度不言而喻。与塞利尼索同靶点的药物还包括Karyopharm Therapeutics公司的艾他尼索(Eltanexor), 用于治疗骨髓增生异常综合征、前列腺癌、结直肠癌和多发性骨髓瘤, 目前处于临床Ⅱ期; Karyopharm Therapeutics公司的管线中还有处于临床Ⅰ期研究的凡迪尼索(Verdinexor)和BIIB-100, 分别用于流感病毒感染和肌萎缩性脊髓侧索硬化症; CanBas和Stemline公司的S-03747目前处于临床Ⅰ期, 用于实体瘤的治疗; 辉瑞公司的Leptomycin B通过靶向XPO1治疗细菌感染和实体瘤, 但终止于临床Ⅰ期。
1.2 研发过程Karyopharm Therapeutics公司成立于2008年, 专注于研发核转运体及相关靶标的首创性药物, 并将其用于治疗癌症和其他重大疾病, 具有极强的创新性。其管线中的多个小分子抑制剂可以高效靶向于肿瘤细胞中过表达的XPO1, 正在进行多种肿瘤治疗的临床试验。塞利尼索(Selinexo, 研发代号: KPT300)是Karyopharm Therapeutics公司获批的首个XPO1抑制剂, 为XPO1后续的抑制剂研究奠定了坚实的基础。
由于塞利尼索的分子发现和优化过程尚未有文章发表, 本文仅从相关专利中推断其抑制剂发现和优化过程, 与真实研发情况或有出入。Karyopharm Therapeutics公司针对XPO1靶标已发现具有多种化学结构的小分子, 塞利尼索是以图 2中的化学骨架(chemical scaffold)优化而来, 此类结构母核的抑制剂均具有较好的骨髓瘤细胞抑制活性。在分子优化过程中, 如果改变化学骨架苯环上的取代基, 例如替换其中一个三氟甲基(化合物1、2)或者添加卤素(化合物3), 虽然仍具有较高的细胞抑制活性(IC50 < 100 nmol·L-1), 但其血药浓度较低, 不适宜继续开发, 证明苯环骨架间位的双三氟甲基取代最优。通过研究与肼相连的芳环构效关系, 发现多种取代基的替换可以实现血药浓度的提高, 例如化合物4, 其AUC值高达12 300 h·ng·mL-1, 但其存在B:P值过高(5.0)的问题, 可能会导致药物在脑部蓄积产生毒性。最后通过多种芳杂环等取代基的尝试, 发现了吡嗪取代后的化合物6 (Selinexor)细胞抑制活性保持, 且血药浓度明显提高, B:P值也显著降低(0.71), 证明此化合物在活性保持的同时, 具有较优的药代动力学性质。构效关系与成药性优化的过程中还发现, 若将肼结构中的一个氨基甲基化后(化合物5), 其药效也大幅降低。免疫印迹实验表明, 浓度为10~ 50 nmol·L-1的Selinexor即可在细胞内高效抑制XPO1, 并显著影响相关肿瘤抑制蛋白的表达(Patent No.: US010544108B2)。基于Selinexor进行研究发现, XPO1通过与真核细胞起始因子4E形成复合物, 参与到细胞周期相关蛋白(细胞周期启动子、cyclin D1、cyclin E、CDK2/4/6)和抗凋亡蛋白(Mcl-1、Bcl-xL)的生物学功能中[7-9]。此外, Selinexor可以抑制RANKL诱导的NF-κB和NFATc1表达, 直接损害破骨细胞形成和骨吸收, 对成骨细胞和BMSCs影响极低[6, 10, 11]。以上广泛的蛋白表达和基因转录变化的综合结果, 最终实现了Selinexor的细胞周期阻滞和肿瘤细胞凋亡的功能。

|
Figure 2 The discovery and development of Selinexor (6). (B: P refers to brain to plasma; NT: Not tested) |
塞利尼索通过结合并抑制核输出蛋白XPO1发挥作用, 导致肿瘤抑制蛋白在细胞核内积累并重新启动, 这一过程会放大它们的肿瘤抑制功能, 导致肿瘤细胞死亡, 同时不会对正常细胞造成显著影响。塞利尼索作为孤儿药, 其受众相对较小, 该药用于已经接受过至少两种蛋白酶体抑制剂或两种免疫调节剂及CD38单抗药物治疗后仍无效的难治型多发性骨髓瘤。塞利尼索的此次新药申请是基于一项临床Ⅱb期的STORM的数据结果, 该研究中招募了122例已接受过度治疗且对蛋白酶体抑制剂、免疫调节剂和CD38抗体均耐药的难治型多发性骨髓瘤患者。患者接受塞利尼索(80 mg, 口服, 每周2次)联合低剂量地塞米松(20 mg, 每周2次)进行治疗, 主要疗效终点ORR (总缓解率)达到25.3%, 起效时间的中位数为4周, 持续时间的中位数为3.8个月。截止目前, 已经有超过2 500名患者接受了此治疗方案, 塞利尼索的成功为广大无药可医的难治型多发性骨髓瘤患者带去了福音[10]。
此外, 塞利尼索还有多个适应症处于临床阶段, 其中治疗脂肪肉瘤和子宫内膜癌的临床研究处于Ⅲ期阶段, 转移性乳腺癌、小细胞肺癌、转移性前列腺癌、胶质母细胞瘤、鳞状细胞癌、弥漫性大B细胞淋巴瘤、骨髓异常增生等多项研究均处于不同的临床阶段。除了塞利尼索, Karyopharm公司管线中针对XPO1靶标还有多款抑制剂处于临床研究。由此可见, 首创性药物的成功研发往往会带来巨大的临床应用和开发潜力[9, 12]。
2 替那帕诺(Tenapanor)——全球首个靶向于Na+/H+交换器NHE3的IBS-C治疗药物 2.1 研发背景肠易激综合征(irritable bowel syndrome, IBS)是一种持续或间歇性发作的肠道功能紊乱行疾病, 其主要症状为腹痛、腹胀及腹部不适, 常伴有排便习惯、排便频率和排便性状的改变。通常根据其异常排便的习惯进行分类, 主要包括便秘型(IBS-C)、腹泻型(IBS-D)、混合型(IBS-M)和不定型(IBS-U)。其中, IBS-C的具体起因未知, 无合适的诊断方法和生物标志物。当前美国的IBS-C患者超过1 000万人, 基数庞大, 严重影响着人们的生活质量和生命健康[13]。
NHE3 (sodium-hydrogen exchanger type 3)属于NHE基因家族, 主要存在于肠上皮细胞的顶膜、脑干细胞区和肾脏组织中, 其作为Na+/H+交换器的主要功能是介导近端小管Na+离子再吸收[14-16]。在胃肠道中, NHE3正常生理状态下维持胃肠道内外的Na+/H+离子平衡(图 3)。若能选择性抑制NHE3, 会降低NHE3的离子交换功能, 减少饮食中的Na+离子吸收, 导致肠道内的Na+离子蓄积, 最终通过增加肠道内的液体含量而软化大便, 减轻便秘的症状[17]。

|
Figure 3 Biological mechanism of NHE3 and inhibition effects |
在替那帕诺上市之前, IBS-C的临床治疗药物主要为各类泻药、促动力剂和促分泌剂。FDA批准的用于治疗IBS-C的药物包括利那洛肽(Linaclotide)[18]、替加色罗(Tegaserod)[19, 20]和芦比前列酮(Lubiprostone)[21]。然而, 这些药物都具有明显的缺陷:利那洛肽是全球首个鸟苷酸环化酶-C受体激动剂, 在治疗便秘的药物领域处于领先地位, 但是利那洛肽不适用于年纪小于17岁的患者; 替加色罗是一款选择性血清素-4受体激动剂, 也是此靶点唯一被批准用于IBS-C治疗的药物, 但存在潜在的心血管安全问题, 极大地限制了此药物的应用; 芦比前列酮是一款氯离子通道激活剂, 2008年被FDA批准用于女性的IBS-C患者, 占据了一定的市场份额。2019年9月, 替那帕诺作为全球首款NHE3抑制剂获批上市, 可以通过全新的作用机制为IBS-C患者提供了更多的治疗选择。
2.2 研发过程替那帕诺(研发代号: AZD-1722; RDX5791)是由Ardelyx公司研发的一种高选择性的、强效的NHE3抑制剂, 对NHE3的抑制活性IC50值为5 nmol·L-1, 对人肠道转运蛋白NHE1 (SLC9A1)、NHE2 (SLC9A2)、TGR5 (GPBAR1)、ASBT以及Pit-1均无抑制活性。替那帕诺对于钠离子和磷酸盐吸收的抑制作用是选择性的, 不会影响肠道吸收其他离子、分子和营养物质, 可以保证肠道的正常营养摄取。此外, 由于分子特定的理化性质, 药物本身也不会被胃肠道吸收, 而是会随排泄物排出体外, 降低了不良反应发生的可能性。与传统药物研发不同的是, 考虑到IBS-C的发病机制与胃肠道特殊的功能结构, 通过靶向NHE3的治疗IBS-C的抑制剂需具备以下几个特点: ①低生物利用度(较少被胃肠道吸收, 降低毒副作用); ②弱透膜性(较少被上皮细胞吸收, 利于药物排出); ③高选择性和胃肠道蓄积效应(不影响其他组织器官中的NHE3)。
为了获得满足以上条件的NHE3抑制剂, Ardelyx公司在研发起始阶段首先选定了3类具有NHE抑制功能的单价化合物(monovalent) (参见图 4的“Chemical Scaffold A-C”), 他们均具有一定的NHE蛋白抑制活性, 但是其化合物分子量小、生物利用度较高、易透膜, 不满足上述的抑制剂设计要求。通过合理增加化合物的分子量和极性表面积(tPSA)可以有效降低分子的生物利用度和透膜性, 因此第二阶段的研究集中于NHE多价化合物(polyvalent)。通过在“Chemical Scaffold A”的骨架中引入不同的长链, 逐步探索适宜设计多价化合物的位点。如图 4所示, 化合物7相较其他化合物在此阶段表现出良好的性质, 在保持良好抑制活性的同时, 口服给药后血药浓度低(AUC = 53 ng·h·mL-1, 2.1 mg·kg-1口服), 膜透过性较弱[Avg Papp = 0.53 cm·(μs)-1], 证明可以通过此策略建立多价化合物。第三阶段的研究聚焦于探索最优的“Core”结构。通过尝试替换各类“Core”连接链结构, 最终得到的化合物10即为替那帕诺, 其在保持优异的抑制活性(IC50值为低纳摩尔级别)的同时, 具有适宜的拓扑极性表面积(tPSA = 235 Å)。替那帕诺在体内的肠道蠕动能力恢复实验测试中表现优异(给药15 mg·kg-1可恢复超过40%的肠道蠕动能力), 并在60 min的跨膜电阻测试中表现出不易渗透至胃肠道表皮的特性(Patent No.: US010376481B2)。

|
Figure 4 The discovery and development of Tenapanor (10) |
从替那帕诺的研发过程可以发现, 针对首创性的药物靶标设计抑制剂时, 需要根据疾病的机制特征进行药物的合理设计。该药物设计中, 为了满足特定的治疗需要, 甚至通过降低分子的生物利用度、降低药物的透膜性等策略获得有效的抑制剂。替那帕诺的发现满足了上述所有基于NHE3抑制机制实现IBS-C治疗的苛刻要求, 以一种“非典型”的研发思路获得了有效的药物分子。
2.3 治疗应用NHE3主要表达于小肠和结肠, 促进吸收食物中的钠离子。替那帕诺通过高效抑制NHE3分子, 阻止胃肠道上皮细胞对钠离子的重吸收, 使其积聚于胃肠道内, 进而促进机体向胃肠道内腔分泌水分, 实现软化大便、帮助排便的目的。替那帕诺的Ⅲ期临床试验招募了593名满足标准的IBS-C患者, 以每天两次, 每次50毫克的剂量服用替那帕诺。与安慰剂组相比, 在12周时间内, 至少有6周替那帕诺给药组的患者腹痛明显减弱。同时, 在一周的给药时间内, 接受治疗的患者至少可以增加一次自然完成的排便, 替那帕诺最终通过26周具有统计学意义的研究成功获批上市。
此外, NHE3还广泛分布于肾脏组织中, 替那帕诺还有一项治疗终末期肾病(ESRD)和高磷血症的研究正处于临床Ⅲ期, 有望作为新适应症申请上市。与替那帕诺同靶点的抑制剂还有Sigma-Tau公司的罗他福辛(Rostafuroxin), 临床适应症为高血压, 可以同时靶向于NHE1/2/3/5, 目前正处于临床Ⅱ期。赛诺菲公司研发的AVE-0657也可以靶向于NHE3, 临床适应症为睡眠呼吸暂停综合征和呼吸道疾病, 但由于有效性较差而终止于临床Ⅱ期。由于IBS-C的患者众多, 替那帕诺作为全球首个治疗IBS-C的NHE3抑制剂, 预计销售峰值可达4~5亿美金。
3 拉米地坦(Lasmiditan)—全球首款用于治疗偏头痛的5-羟色胺1F受体激动剂药物 3.1 研发背景偏头痛是一种常见的原发性头痛, 常表现为单侧的搏动性头痛, 发病时还伴有恶心、呕吐等症状, 是神经内科最常见的疾病之一。在全世界范围内, 偏头痛都严重影响着人们的生活质量。根据FDA的数据显示, 在美国有超过3 000万的偏头痛患者, 2%的美国人每月中有超过一半的时间受到偏头痛的困扰, 造成的社会经济负担为10~17亿美元。偏头痛属于复杂的神经疾病, 发病机制复杂, 尚无明确的作用机制和有效的治疗药物[22]。
5-羟色胺受体(5-HT, 又称血清素受体)是一大类存在于中枢神经中央和末梢神经系统的G蛋白偶联受体及配体门控离子通道, 负责调控兴奋性和抑制性的神经传导物质。5-HT家族庞大, 包括1~7七个亚科, 其中5-HT1是种类最多的一类亚科。现有的治疗药物主要为“曲坦”(-triptan)类药物, 主要靶标为5-HT1B/1D, 但其治疗效果较差, 部分患者存在复发情况, 且对中重度患者的疗效显著降低。5-HT1B/1D受体激活会引起曲坦类药物的血管收缩不良反应, 不适用于心脑血管和周围血管有疾病的患者, 因此亟需研发具有全新作用机制的治疗药物。5-羟色胺1F受体(5-HT1F)主要表达于脑部、子宫和肠系膜, 广泛分布于三叉神经网络系统、新大脑皮层、小脑及海马体中。如图 5所示, 有研究表明突触前5-HT1F受体的激活可以抑制降钙素基因相关肽(CGRP)的信号释放, 从而阻断三叉神经尾核内部的神经元信号传导, 产生抗偏头痛的作用[23-26]。

|
Figure 5 Biological mechanism of 5-HT1F receptor agonist for the treatment of migraine |
2019年10月, FDA批准了礼来公司研发的拉米地坦, 是全球首款也是唯一的5-HT1F受体激动剂, 属于新型的地坦类药物。拉米地坦通过高亲和力与5-HT1F受体结合后激活其功能, 高效抑制CGRP的信号释放并阻断神经元信号传导, 产生抗偏头痛的治疗效果[27]。由于人体血管表面尚未发现CGRP受体, 因此拉米地坦无血管收缩作用, 适用于不能服用曲坦类药物的高血压及心脏病的患者, 且疗效优于曲坦类药物[24]。
3.2 研发过程在拉米地坦发现之前, 治疗偏头痛的药物主要为曲坦类的5-HT1B/1D受体激动剂, 例如夫罗曲坦(Frovatriptan)和依来曲坦(Eletriptan)均为低纳摩尔级别的5-HT1B/1D受体的激动剂, 通过阻断神经末梢的信号传导而起效(图 6)。然而, 激活5-HT1B/1D受体对偏头痛患者的响应率较低, 并且会引起严重的血管收缩不良反应, 不适用于具有潜在的心血管风险的偏头痛患者。为了解决这一问题, 研究发现激活5-HT1F受体可以选择性地抑制CGRP信号释放, 阻断痛觉传递, 并利用血管表面无CGRP受体这一特点, 巧妙地从生理机制上避免此类药物的心血管不良反应的产生。因此, 第一阶段寻找5-HT1F受体激动剂的工作基于曲坦类药物的吲哚类结构母核, 通过一系列结构改造后得到化合物LY334370, 其5-HT1F受体激动活性达到1.87 nmol·L-1。然而, 由于其母核结构仍为吲哚, 不可避免地对5-HT1A/1B/1D等受体均具有较强的激动活性, 选择性较差, 难以克服曲坦类药物引起心血管风险的不良反应。第二阶段的研究目标需要突破曲坦类药物的母核结构, 得到具有全新结构类型的5-HT1F受体激动剂。如图 6所示, Eli Lilly公司研发过程中发现一类具有吡啶母核结构的化合物可以激活5-HT1F受体, 通过深入的构效关系探索最终得到的拉米地坦实现了化学结构母核的突破, 其化学结构完全不同于曲坦类药物, 是一种高选择性的5-HT1F受体激动剂(Ki = 2.1 nmol·L-1), 对同源的5-HT1B/1D受体无活性(Ki > 1 000 nmol·L-1), 选择性超过470倍(Patent No. US8748459B2)[28]。

|
Figure 6 The discovery and development of Lasmiditan |
拉米地坦的主要疗效确证基于一项Ⅲ期临床试验结果。为了证明拉米地坦对成人急性偏头痛治疗的安全性和有效性, 共招募了3 177名有偏头痛病史的患者。实验结果表明, 服用拉米地坦后2 h偏头痛患者症状减轻的比例显著提高, 同时明显减弱患者的恶心、声光敏感等症状。拉米地坦的优势还在于未表现出血管收缩活性, 受用人群范围更广, 且对曲坦类药物不敏感的患者具有良好的效果, 不良反应较少[29]。与拉米地坦同靶点的药物还有礼来公司的LY- 334370, 适应症同为偏头痛, 正处于临床Ⅱ期研究中。
拉米地坦是一个全新的、首创性分子, 是过去20年中出现的针对偏头痛治疗的首个具有新作用机制的药物, 研发和市场费用接近10亿美元, 耗时超过15年。拉米地坦的成功上市不仅证明了5-HT1F受体激动剂作为偏头痛的治疗策略可行, 也为偏头痛患者提供了新的治疗选择, 尤其是伴有高危心血管风险因素的偏头痛患者群体, 具有巨大的市场价值[27]。
4 结语与展望首创性药物的研发对工业界和学术界都极具吸引力, 不仅因为首创性药物可以率先占领市场取得可观的经济收益, 其研究成果也可以彰显研究团队的研发实力, 成为全新药物靶标的奠基者和领路人。不可忽视的是, 首创性药物研发的超高风险与投入, 令许多制药巨头折戟沉沙, 也令大多数研究团队望而却步。基于全新靶标的首创性药物研发需要对靶标的生物机制、结构特征与生理功能有全面的了解, 并以此为基础开展调控剂的筛选、结构改造、成药性优化等后续研究。本文所述的3款药物均为本年度首创性药物, 其研究目的与研发思路也各不相同。塞利尼索的成功依靠Karyopharm Therapeutics公司对核转运体及相关靶标的专注研究, 实现了对此类全新靶标的突破, 有望应用于多种重大疾病的治疗中。替那帕诺的成功依靠NHE3在IBS-C治疗中的独特机制, 通过合理设计低生物利用度、低透膜性的多价NHE3抑制剂实现其治疗目的。拉米地坦的成功依靠对5-HT1F受体的选择性激动作用, 以全新机制克服曲坦类药物的不良反应, 扩大了适用患者的范围。本文通过浅析2019年FDA批准的3款具有一定代表性的首创性小分子药物的研究背景、研发过程及治疗应用, 为更多首创性药物的研究提供少许借鉴与思考。
2020年新冠病毒席卷全球, 严重威胁着人们的生命健康。面对未知病毒的威胁, 需要更加夯实药物研发的技术手段, 优化药物研发的全流程, 以解决切实的疾病需求为出发点, 创制更多普惠大众的首创性药物。
作者贡献:王磊负责文章的资料收集与撰写; 尤启东负责文章的先题与修改, 为该文章的主要负责人。
利益冲突:本文无利益冲突。
| [1] |
Guo Z. Concise analysis for innovation of pioneering and follow-on drugs[J]. Acta Pharm Sin (药学学报), 2016, 51: 1179-1184. |
| [2] |
Karyopharm Announces FDAApproval of XPOVIO™ (selinexor)for the Treatment of Patients with Relapsed or Refractory Multiple Myeloma [EB/OL]. https://investors.karyopharm.com/newsreleases/news-release-details/karyopharm-announces-fda-approval-xpoviotm-selinexor-treatment.
|
| [3] |
Fukuda M, Asano S, Nakamura T, et al. CRM1 is responsible for intracellular transport mediated by the nuclear export signal[J]. Nature, 1997, 390: 308-311. DOI:10.1038/36894 |
| [4] |
Nguyen KT, Holloway MP, Altura RA. The CRM1 nuclear export protein in normal development and disease[J]. Int J Biochem Mol Biol, 2012, 3: 137-151. |
| [5] |
Turner JG, Sullivan DM. CRM1-mediated nuclear export of proteins and drug resistance in cancer[J]. Curr Med Chem, 2008, 15: 2648-2655. DOI:10.2174/092986708786242859 |
| [6] |
Tai YT, Landesman Y, Acharya C, et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma:molecular mechanisms and therapeutic implications[J]. Leukemia, 2014, 28: 155-165. DOI:10.1038/leu.2013.115 |
| [7] |
Chari A, Vogl DT, Gavriatopoulou M, et al. Oral selinexor-dexamethasone for triple-class refractory multiple myeloma[J]. N Engl J Med, 2019, 381: 727-738. DOI:10.1056/NEJMoa1903455 |
| [8] |
Zhong Y, El-Gamal D, Dubovsky JA, et al. Selinexor suppresses downstream effectors of B-cell activation, proliferation and migration in chronic lymphocytic leukemia cells[J]. Leukemia, 2014, 28: 1158-1163. DOI:10.1038/leu.2014.9 |
| [9] |
Etchin J, Montero J, Berezovskaya A, et al. Activity of a selective inhibitor of nuclear export, selinexor (KPT-330), against AML-initiating cells engrafted into immunosuppressed NSG mice[J]. Leukemia, 2016, 30: 190-199. |
| [10] |
Syed YY. Selinexor:first global approval[J]. Drugs, 2019, 79: 1485-1494. DOI:10.1007/s40265-019-01188-9 |
| [11] |
Neggers JE, Vercruysse T, Jacquemyn M, et al. Identifying drug-target selectivity of small-molecule CRM1/XPO1 inhibitors by CRISPR/Cas9 genome editing[J]. Chem Biol, 2015, 22: 107-116. DOI:10.1016/j.chembiol.2014.11.015 |
| [12] |
Bhatnagar B, Zhao Q, Mims AS, et al. Selinexor in combination with decitabine in patients with acute myeloid leukemia:results from a phase 1 study[J]. Leuk Lymphoma, 2020, 61: 387-396. DOI:10.1080/10428194.2019.1665664 |
| [13] |
Fichna J, Wood JT, Papanastasiou M, et al. Endocannabinoid and cannabinoid-like fatty acid amide levels correlate with pain-related symptoms in patients with IBS-D and IBS-C:a pilot study[J]. PLoS One, 2013, 8. |
| [14] |
Yun CH, Oh S, Zizak M, et al. cAMP-mediated inhibition of the epithelial brush border Na+/H+ exchanger, NHE3, requires an associated regulatory protein[J]. Proc Natl Acad Sci U S A, 1997, 94: 3010-3015. DOI:10.1073/pnas.94.7.3010 |
| [15] |
Donowitz M, Cha B, Zachos NC, et al. NHERF family and NHE3 regulation[J]. J Physiol, 2005, 567: 3-11. DOI:10.1113/jphysiol.2005.090399 |
| [16] |
Donowitz M, Mohan S, Zhu CX, et al. NHE3 regulatory complexes[J]. J Exp Biol, 2009, 212: 1638-1646. DOI:10.1242/jeb.028605 |
| [17] |
Honegger KJ, Capuano P, Winter C, et al. Regulation of sodium-proton exchanger isoform 3(NHE3) by PKA and exchange protein directly activated by cAMP (EPAC)[J]. Proc Natl Acad Sci U S A, 2006, 103: 803-808. DOI:10.1073/pnas.0503562103 |
| [18] |
McCormack PL. Linaclotide:a review of its use in the treatment of irritable bowel syndrome with constipation[J]. Drugs, 2014, 74: 53-60. DOI:10.1007/s40265-013-0157-5 |
| [19] |
Liem O, Mousa HM, Benninga MA, et al. Tegaserod use in children:a single-center experience[J]. J Pediatr Gastroenterol Nutr, 2008, 46: 54-58. DOI:10.1097/01.mpg.0000304454.99799.42 |
| [20] |
Thompson CA. Novartis suspends tegaserod sales at FDA's request[J]. Am J Health Syst Pharm, 2007, 64: 1020. |
| [21] |
McKeage K, Plosker GL, Siddiqui MA. Lubiprostone[J]. Drugs, 2006, 66: 873-879. DOI:10.2165/00003495-200666060-00015 |
| [22] |
Curto M, Cipolla F, Cisale GY, et al. Profiling lasmiditan as a treatment option for migraine[J]. Expert Opin Pharmacother, 2020, 21: 147-153. DOI:10.1080/14656566.2019.1694004 |
| [23] |
Vila-Pueyo M. Targeted 5-HT1F therapies for migraine[J]. Neurotherapeutics, 2018, 15: 291-303. DOI:10.1007/s13311-018-0615-6 |
| [24] |
Hougaard A, Tfelt-Hansen P. Review of dose-response curves for acute antimigraine drugs:triptans, 5-HT1F agonists and CGRP antagonists[J]. Expert Opin Drug Metab Toxicol, 2015, 11: 1409-1418. DOI:10.1517/17425255.2015.1055244 |
| [25] |
Olesen J. 5-Hydroxyptryptamine 1F (5-HT1F) receptor agonism[J]. Cephalalgia, 2010, 30: 1157-1158. DOI:10.1177/0333102410377671 |
| [26] |
Mitsikostas DD, Tfelt-Hansen P. Targeting to 5-HT1F receptor subtype for migraine treatment:lessons from the past, implications for the future[J]. Cent Nerv Syst Agents Med Chem, 2012, 12: 241-249. DOI:10.2174/187152412803760627 |
| [27] |
Lamb YN. Lasmiditan:first approval[J]. Drugs, 2019, 79: 1989-1996. DOI:10.1007/s40265-019-01225-7 |
| [28] |
Nelson DL, Phebus LA, Johnson KW, et al. Preclinical pharmacological profile of the selective 5-HT1F receptor agonist lasmiditan[J]. Cephalalgia, 2010, 30: 1159-1169. DOI:10.1177/0333102410370873 |
| [29] |
Goadsby PJ, Wietecha LA, Dennehy EB, et al. Phase 3 randomized, placebo-controlled, double-blind study of lasmiditan for acute treatment of migraine[J]. Brain, 2019, 142: 1894-1904. DOI:10.1093/brain/awz134 |