 2020, Vol. 55
2020, Vol. 55

新药创制是复杂的智力活动,涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹,而构建化学结构是最重要的环节,因为它涵盖了药效、药代、安全性和生物药剂学等多维性质。本栏目以药物化学视角,对有代表性的药物的成功构建,加以剖析和解读。
泽布替尼是是我国本土研发成功的抗肿瘤新药。作为跟随性药物,虽然省去了靶标的概念验证,但应在安全有效性能上优于先行者。本品的研发目标明确,临床前数据表明达到了提高选择性和生物利用度的目标。构建的分子结构是在全新的母核骨架上安排、调整和优化结构,走的是创新之路。在时间轴上,从2012年立项到2019年底FDA批准上市,七年时间成功地创制了国际水平的新药,质量和效率都是很高的。本文从药物化学视角简要地叙述了研发的脉络,希望对国内新药创制有所启示和示范。
(编者按)
布鲁顿酪氨酸激酶(BTK)是B细胞受体信号通路的重要组成部分, 在B淋巴细胞的发育和生长中扮演重要角色。当BTK基因由于突变而失活时, 骨髓中的B细胞就无法发育成熟。若过度活跃, 同样会带来病变, 白血病和淋巴瘤患者体内的癌细胞中, BCR信号通路经常处于异常激活的状态, BTK蛋白的水平提高, 因而BTK是研制抗癌药物的靶标。
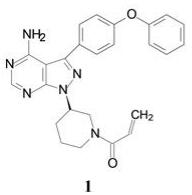
1.2 市场状态我国百济神州医药公司2012年立项研发BTK抑制剂, 当时国外已有处于临床试验的候选物, 还没有批准上市的药物。2013年Pharmacyclics和强生公司首创的伊鲁替尼(1, ibrutinib)上市(Pan Z, Scheerens H, Li S, et al. Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. ChemMedChem, 2007, 2: 58-61), 2017年阿斯利康公司的阿卡替尼(2, acalabrutinib)是第二个上市的BTK抑制剂(Wu J, Zhang MZ, Liu DL. Acalabrutinib (ACP-196): a selective second-generation BTK inhibitor. J Hematol Oncol, 2016, 9: 21)。本品启动研究时, BTK靶标已经临床试验的概念验证, 确证为可药性靶标, 因而是个跟随性的新药创制项目。
|
|
研制跟随性药物设定的产品目标须在安全有效上优胜于首创药物。分析当时在研的项目, 研发者将提高口服吸收性和选择性作为目标, 是针对伊鲁替尼的不足, 即口服生物利用度低(F < 10%), 剂量偏大, 治疗窗口窄, 而且对野生型表皮生长因子受体(EGFR)呈现抑制, 引起皮疹和腹泻等不良反应。所以提高口服吸收性和选择性, 可降低用药剂量, 扩大治疗窗口, 减少不良反应。目标物的化学结构特征为不可逆结合, 以期药物作用的持久性和减低用药频度。
2 活性评价 2.1 生化测定化合物对BTLK抑制活性重组的BTK蛋白与受试物温孵后, 加入ATP和肽底物(Biotin-AVLESEEELYSSARQ-NH2), 加入含有隐式铕(europium cryptate)缀合的p-Tyr66抗体和链霉亲和素标记的XL665的终止液以终止反应。用BMG PHERAstar FS仪器测定时间分辨荧光共振能量转移(TR-FRET)信号强度, 计算化合物的IC50, 数值越小活性越高。
2.2 生化测定对ITK/TEC/JAK3/EGFR/HER2的作用受试物对这些酶的抑制活性也是用TR-FRET方法测定, 计算化合物的IC50, 数值越大化合物脱靶作用越小。
2.3 生化测定对细胞中BTK的抑制活性用基于均相时间分辨荧光法(HTRF)定量测定受试物对高表达BTK的Ramos细胞中BTK Tyr-223的磷酸化作用。Ramos细胞与不同浓度的受试物温育3 h, 加入过钒酸钠, 使细胞溶解, 一定量的细胞溶解液中加入anti-BTK-d2和anti-pBTKK抗体混合液, 暗处放置过夜, 用兼容性HTRF读出在两个波长(665、620 nm)下的荧光发射强度, 根据对这两个信号强度的抑制比计算化合物对细胞抑制的活性强度。
2.4 脱靶性评价对Rec-1和OCI-LY10细胞的活力、EGFR pY1068的细胞活性、Jurkat细胞的ITK pPLCγ1活性和对高表达HEK293细胞的抑制活性测定都是为了评价化合物在细胞水平上的选择性作用, 数值越大脱靶作用越小。
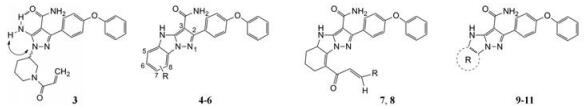
3 先导物的骨架演化 3.1 母核氨基咪唑的结构变换作为跟随性药物, 研制者的理念也是以竞争激酶ATP结合腔为作用模式, 抑制剂模拟ATP腺嘌呤环, 并变换杂环的结构骨架, 还借鉴1和2都在嘌呤的7位有疏水性芳环的结构特征, 设计了氨基咪唑酰胺类型的化合物, 酰氨是为了形成分子内氢键, 用假杂环模拟平面性稠合的嘧啶环(Wang ZW, GuoYH. Protein kinase inhibitors and uses thereof. US Patent US2015005277, 2015-01-01)。在适宜的位置连接迈克尔加成基团, 以实现不可逆抑制。在一系列化合物中发现化合物3具有活性, R和S异构体对BTK的抑制活性IC50值分别为0.21和0.80 nmol·L-1, 抑制BTK Tyr-223细胞活性IC50值分别为1.0和3.8 nmol·L-1, 然而对EGFR的抑制活性也很高, 选择性差意味着有脱靶作用。共晶结构显示3 (无N-丙烯酰基)的结合模式与伊鲁替尼的结合模式相似(图略)。
进而将氨基与吡唑环并合设计了吡唑并苯并咪唑为骨架的化合物如4~6, 没有丙烯酰基的化合物4活性不高, 处于7-位的丙烯酰胺取代(5)活性也很弱, 而8位的活性(6)显著提高。将6的苯环部分饱和为7和8 , 进一步提高了活性, 将迈克尔基团自8位移至7位的化合物9提高了细胞活性, 但扩环成䓬环的10和11活性未见提高。表 1列出了上述母核演化过程的结构和活性。
| Table 1 Structures and selected properties of 3-11 |
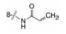
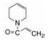
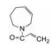
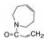
化合物4~11的母核为三环稠合, 刚性较强不利于药代性质, 为此将苯环或氮杂环由并合改为单键连出, 合成的代表性化合物列于表 2。结果表明, 双环母核体系只要有丙烯酰胺的”弹头”仍然保持活性, 但连接的位置很重要, 例如13和15的抑酶活性显著高于12和14, 这是因为与Cys481的位置合适的缘故。若将Cys481突变成Ser481则活性降低, 说明迈克尔加合基团与巯基结合对活性的重要性。13对EGFR活性很低, 较少脱靶作用, 但它抑制细胞活性不如15, 不过15的药代性质有缺陷, 小鼠一次剂量10 mg·kg-1, 4 h后脾细胞的占有率只有56%, 所以仍然有待优化新的母核。
| Table 2 Structures and selected properties of the compounds with bicyclic core |
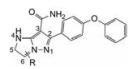
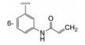
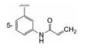
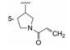
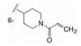
由于变换母核合成的吡唑并吡啶(未列出)的化合物活性很低, 推测与骨架的过于刚性有关, 遂变换母核为吡唑并哌啶, 部分饱和化降低了分子的平面性, 或许有利于结合。合成的代表性化合物列于表 3。化合物16的一对对映体(16a和16b, 拆分了但未确定绝对构型)对BTK酶和细胞活性相近, 选择性也比较高。而将迈克尔基团移至对位, 活性显著下降。然而连接在邻位的化合物18a和18b的活性差别很大, 活性相差300多倍, 提示处于邻位的迈克尔基团与Cys481结合的立体选择性非常高。18a的细胞活性和选择性都非常高, 而N-甲基化的化合物19的活性陡然下降, 提示这个位置的空间配置是非常严格的。这几个高活性的化合物的大鼠药代动力学表明口服生物利用度很低(例如18a的F值只有1.7%), 因而不值得深入研究。不过以吡唑并哌啶为母核的骨架, 连接的苯环邻位有迈克尔侧链的结构是个优化的配置。
| Table 3 Structures and selected properties of compounds with pyrrazolopiperidine core |
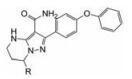

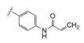
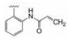
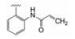
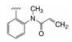
下一步是固定18a的下半部分, 变换二苯醚部分, 合成的代表性化合物列于表 4。将末端的苯基用较亲水性的四氢呋喃(20)或环丙甲基(21)替换, 抑酶活性基本不变, 20对细胞活性减弱, 可能是过膜性减弱所致。对EGFR的抑制作用也基本没变。苯氧基换作体积较小的基团如甲氧基、甲基或氯原子, 活性仍在纳摩尔水平。化合物21、23和27的溶解性强于18a, 对肝脏微粒体的代谢稳定性也优于18a, 灌胃大鼠的生物利用度也高于18a, 只是所有这些化合物对EGFR仍显示强抑制活性, 预示会有不良反应, 须要继续优化。
| Table 4 Structures and properties of pyrrazolopiperidine series with varied phenyl ether part |
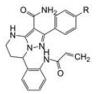
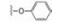

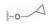

为了提高抑制BTK的选择性抑制、降低对EGFR的作用和提高化合物的生物利用度, 下一步仍以18a为起点, 变换分子中另一端, 即连接丙烯酰胺的苯环为不同的脂肪环或链状结构。并将手性目标物拆分成光活体, 测定必要的活性数据, 结果列于表 5中。化合物29、30和31的活性与18a相当, 而32和33的活性显著降低, 提示环丙基和偕二甲基的位阻效应妨碍了发生共价结合反应。29、30和31的消旋体对BTK激酶都表现高抑制活性, 拆分成光学异构体, 分别测定对BTK、EGFR和细胞活性, 提示29a的活性和选择性略优于30a和31a, 需要作进一步比较。
| Table 5 Structures and activity of the compounds with varied carrier of acrylamide moiety |
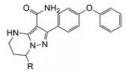
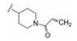
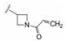
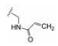
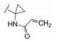
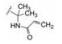
化合物29a、30a和31a显示为高活性化合物, 用大鼠静脉注射和灌胃比较药代性质, 结果列于表 6, 表明化合物29a的半衰期、清除率、tmax、Cmax、AUC和口服生物利用度都优于30a和31a, 从而确定29a为候选化合物。
| Table 6 Pharmacokinetic property of compound 29a, 30a and 31a |
为了确定化合物29a为候选化合物进入开发阶段, 对其血浆蛋白结合率, 人、犬和啮齿动物的肝微粒体清除率, 对重要细胞色素P450的抑制作用以及对小鼠移植性OCI-LY10 DLBCL等肿瘤的体内试验(数据从略), 还与伊鲁替尼对15种激酶的抑制作用作比较, 表明29a对多种酶系的作用优于(或不劣于)伊鲁替尼, 数据列于表 7中。
| Table 7 Biochemical kinase selectivity of 29a and ibrutinib |
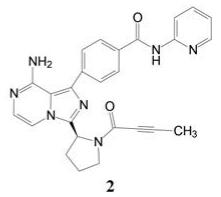
化合物29a定名为泽布替尼(zanubrutinib), 于2014年在包括中国在内的全球进行临床试验, 证明是治疗套细胞淋巴瘤和慢性淋巴性白血病/小淋巴细胞淋巴瘤的有效药物, 于2019年经FDA批准上市。
6 泽布替尼与BTK激酶结合模式泽布替尼与BTK激酶共晶结构显示, 分子定位于ATP结合腔内, 酰氨基与铰链的Glu475和Met477形成氢键网络, 哌啶的氮原子也与Met477发生氢键结合, 吡唑的氮原子经水分子(氢键)介导与Lys430的侧链氨基形成氢键, 二苯醚的末端苯环与Phe540发生T-型π-π相互作用, 迈克尔基团与Cys481巯基的距离2.9Å, 处于发生共价结合的范围。此外, 酰氨基与哌啶氮的分子内氢键(图 1中未示)也验证了最初模拟嘧啶酮结构的设想(Guo YH, Liu Y, Hu N, et al. Discovery of zanubrutinib (BGB-3111), a novel, potent, and selective covalent inhibitor of Bruton's tyrosine kinase. J Med Chem, 2019, 62: 7923-7940)。

|
Figure 1 Binding mode of zanubruyinib and BTK from cocrystal structure |
泽布替尼是继依鲁替尼和阿卡替尼之后FDA批准的第三个BTK抑制剂, 是在我国本土研发成功的。作为跟随性药物, 虽然省去了靶标(概念)验证, 但在安全和有效性上应优于依鲁替尼和阿卡替尼, 风险置后是显而易见的。所以设定的目标是提高选择性作用和口服生物利用度, 临床前数据表明达到了预期的目标。就分子结构而言, 乍一看与已上市的两个结构有相似之处, 其实从本文介绍的研发路径看, 某些片段(例如二苯醚和迈克尔基团)有参考的痕迹, 但终究在全新的母核骨架上如何安排、调整和优化结构, 走的是完全创新之路。在时间坐标上, 从立项(2012年)到开始临床研究(2014年), 从设计合成的500多个化合物中优选出了候选物, 开始了临床试验, 到2019年底FDA批准上市, 7年的时间创制了一个国际水平的新药, 可谓高质量高效率的研发。本文从药物化学视角简要地叙述了研发的脉络, 虽然个中的细节和艰辛不得而知, 但对国内业界的新药创制有一定的启示作用。