 2020, Vol. 55
2020, Vol. 55



2. 中国药科大学药学院, 江苏 南京 211198
2. School of Pharmacy, China Pharmaceutical University, Nanjing 211198, China
多梳抑制复合物2 (polycomb repressive complex 2, PRC2)作为多梳家族蛋白(polycomb group of proteins, PcG)的代表性成员, 是一种通过沉默特定基因表达维持核染色质抑制状态的多亚基复合物[1-4]。PRC2的核心部分由组蛋白甲基转移酶enhancer of zeste homologue 2 (EZH2)、胚胎外胚层发育蛋白(embryonic ectoderm development, EED)、suppressor of zeste 12 (SUZ12)组成, 在部分情况下Jumonji and AT-rich interaction domain containing 2 (JARID2)、AE binding protein 2 (AEBP2)等也会出现在PRC2中[5]。PRC2可以催化组蛋白H3赖氨酸27的单、二、三甲基化(H3K27me1、H3K27me2、H3K27me3), 主要过程为:其核心成分和辅因子被招募到目标基因的未甲基化CpG岛, 导致相邻核小体组蛋白H3的27位赖氨酸残基三甲基化(H3K27me3), H3K27me3是PcG相关转录沉默的标志, 通过含有克罗莫结构域(chromo domain)的CBX亚基招募经典的多梳抑制复合物1 (polycomb repressive complex 1, PRC1), 研究认为PRC2与PRC1共同产生基因抑制的作用(图 1)。

|
Figure 1 Polycomb repressive complex 2 (PPRC2) and transcriptional repression |
PRC2的组蛋白赖氨酸甲基转移酶活性主要依赖于复合物中含有SET结构域的EZH2蛋白(图 2)[6], 但是EZH2本身单独存在时既不稳定, 也没有活性。对EZH2催化结构域(520-746)的结构研究表明:单独存在的催化结构域无法识别辅因子S-腺苷-L-甲硫氨酸(S-adenosyl-L-methionine, SAM)和H3K27肽, 这一结果进一步证实了其催化功能不完全[7]。EZH2的甲基转移酶活性至少需要在它与EED以及SUZ12的VEFS结构域(SUZ12-VEFS)的相互作用下才能被刺激产生。低分辨率电子显微镜结构显示SUZ12-VEFS与EZH2的催化结构域紧密靠近, 而EED与EZH2的N末端存在相互作用[8-10]。

|
Figure 2 Structure of PRC2 (PDB ID: 5HYN) |
研究表明PRC2在各种恶性肿瘤、肉瘤、淋巴瘤以及白血病等多种肿瘤疾病的产生、发展以及预后过程中发挥重要的表观遗传学作用(图 3)[11]。该复合物能导致肿瘤抑制基因的甲基化, 从而引起肿瘤的发生和发展。PRC2的关键组成部分EZH2本身也显示出致癌效果[12, 13], 有相关报道揭示EZH2的表达会随着胰腺导管内乳头状黏液性肿瘤的快速生长而增加。最新研究表明EZH2的上调在结直肠癌的进展过程中也起到重要的作用[14]。有报道称EZH2能够控制几种肿瘤细胞的内外耐药性机制, 其中最值得注意的是, 在人类皮肤黑色素瘤中T细胞浸润选择性地与高水平EZH2-PRC2复合物活性相关, 故而在靶向T细胞的免疫疗法过程中, EZH2起到控制黑色素瘤逃逸的分子开关的作用[15]。总之, PRC2活性的功能获得型和功能缺失型改变都可以触发正常组织中的生物学效应, 其活性的上调和下降都可能促进肿瘤发展, 结合“在各种人类癌症中编码PRC2亚基的基因中的体细胞突变存在选择性”这一事实, PcG基因是依赖于环境的致癌基因和抑癌基因[16]。

|
Figure 3 PRC2 related diseases |
有相关研究表明, PRC2在神经元的发育过程中也起到至关重要的作用[17], 它可以防止不当基因(如非神经元基因)的表达[18, 19], 复合物通过甲基化过程阻止分化神经元中转录蛋白到达编码非神经基因的DNA链, 并下调与阿尔茨海默症(Alzheimer's disease, AD)相关的有害基因的表达。近期研究发现, PRC2的表达在某些个体中随着时间的推移而下调[20], 而由于PRC2对上述有害基因的适当调控功能的失效, 发展出迟发性阿尔茨海默症(late-onset Alzheimer's disease, LOAD)的可能性显著增加。
由于PRC2相关蛋白的异常表达显示出了与多种疾病的形成、发展的相关性, 药物化学工作者进行了一些靶向PRC2相关蛋白的小分子调控剂的研究与开发工作, 并取得了一系列进展, 其中, 抑制正常或过度活跃的PRC2可以在几种类型的癌症中降低癌细胞存活率并抑制肿瘤生长[21], 这些结果显示了PRC2抑制剂治疗特定癌症的潜力。本文结合该领域的关注热点以及最新突破, 分两大类对靶向PRC2相关蛋白小分子抑制剂的研究进展进行综述, 为相关研究人员提供参考。
1 靶向EZH2抑制剂(表 1)的研究进展| Table 1 PRC2 complex inhibitors binding to enhancer of zeste homologue 2 (EZH2) protein |
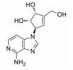
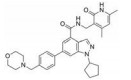
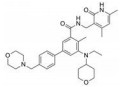
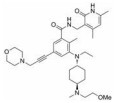
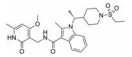
3-脱氮腺嘌呤A (3-deazaneplanocin A, DZNep)是3-脱氮腺苷的一种环戊烯基类似物, 研究发现其可以在乳腺癌细胞中降低EZH2的水平, DZNep也可以抑制其他几种肿瘤细胞的增殖并促进这几种肿瘤细胞的凋亡[22, 23]。作为一种EZH2抑制剂, DZNep可通过细胞G1期阻滞和凋亡从而抑制4种不同类型非小细胞肺癌(non-small cell lung cancer, NSCLC)细胞系的生长, MTT实验显示, DZNep对NSCLC细胞系的抗增殖活性有剂量依赖性, 其IC50范围在0.08~0.24 μmol·L-1, 因此, 以EZH2为靶标的表观遗传学治疗可能是NSCLC的一种潜在有效疗法[24]。通过模拟人生理药代动力学模型(PBPK)的临床前研究表明, 以适当的给药方案静脉注射DZNep可以进一步开发用于NSCLC的治疗[25]。
1.2 吡啶酮类抑制剂EPZ-005687是由Epizyme公司的研究团队于2012年报道的EZH2小分子抑制剂。EPZ-005687抑制PRC2酶活性的IC50值为54 ± 5 nmol·L-1, 相对其他15种蛋白甲基转移酶, EPZ-005687对EZH2有超过500倍的选择性, 而相对于与EZH2密切相关的EZH1, 它对EZH2显示出的选择性也超过50倍[26]。该化合物可降低各种淋巴瘤细胞中的H3K27甲基化水平, 研究结果表明, EPZ-005687对野生型EZH2和Tyr641或Ala677突变型EZH2有同等有效的抑制作用, 这提示该化合物可能在治疗EZH2功能获得型突变相关的疾病方面具有一定的作用[27]。然而, EPZ-005687的体内活性不佳, 这限制了对该化合物的进一步研究。
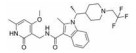
EPZ-6438 (Tazemetostat)也是一个强效高选择性EZH2小分子抑制剂[28], 它与EPZ-005687具有相似的体外活性(Ki=2.5 nmol·L-1, IC50=11 nmol·L-1), 但是药代动力学性质得到了显著提升, 包括在动物实验中显示出的良好的口服生物利用度等。实验表明, 在一系列非霍奇金淋巴瘤(non-Hodgkin lymphomas, NHL)细胞中, 使用EPZ-6438处理可以全局和特异性抑制PRC2底物的甲基化(H3K27)水平, EPZ-6438对具有EZH2突变的NHL细胞具有明显的细胞毒性和抗增殖活性[29]。通过EPZ-6438抑制EZH2可以改变弥漫性大B细胞淋巴瘤(diffuse large B-cell lymphoma, DLBCL)对B细胞激活信号的依赖性[30]。最新的研究结果表明, EPZ-6438与DNA甲基化抑制剂地西他滨联用, 还能够有效上调与脂肪生成相关的PPARγ基因的表达, 促进诱导多功能干细胞来源的间充质干细胞(induced pluripotent stem cell-derived mesenchymal stem cells, iPMSCs)向脂肪细胞分化, 从而显示出新的治疗应用前景[31]。自2016年以来, EPZ-6438在美国获得孤儿药资格, 用于治疗恶性杆状肿瘤(malignant rhabdoid tumors, MRTs)、软组织肉瘤、滤泡性淋巴瘤、间皮瘤和脊索瘤, 并且在具有EZH2激活突变的DLBCL或滤泡性淋巴瘤的治疗中享有快速通道药物特权。2018年, EPZ-6438在欧盟获批成为用于治疗DLBCL、滤泡性淋巴瘤以及恶性间皮瘤的罕见病药物。2019年, Epizyme公司先后向美国FDA递交了Tazemetostat用于治疗不适用于手术切除的转移性或晚期上皮样肉瘤患者、复发或难治性滤泡性淋巴瘤患者的两项上市申请, 其中用于治疗上皮样肉瘤的申请已于2020年1月23日获得了FDA的批准。值得注意的是, 曾有一例患者出现了继发性淋巴瘤的严重不良反应, 导致多项相关临床试验暂停开展。
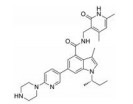
EPZ-011989作为更新的小分子于2015年由Epizyme公司报道, 它是一种高效、选择性、口服有效的EZH2抑制剂, 对野生型EZH2的抑制常数(Ki)小于3 nmol·L-1, 对EZH2具有选择性抑制作用, 与EZH1相比, 选择性大于15倍, 而对其他20种组蛋白甲基转移酶选择性超过3 000倍[32]。在人类B细胞淋巴瘤小鼠异种移植模型中, EPZ-011989表现出明显的肿瘤生长抑制作用。由于EPZ-011989具有非常高的活性、口服生物利用度和代谢稳定性, 且能够显著降低甲基化标记水平并发挥抗肿瘤活性, 它可以作为一种体内工具化合物, 用于进一步研究PRC2复合物在生物学和疾病的临床前模型中的作用。
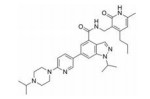
CPI-169也是一种具有代表性的EZH2抑制剂, 可抑制PRC2的催化活性(IC50 < 1 nmol·L-1)并降低H3K27me3在细胞内的水平, 能在多种细胞系中触发细胞周期阻滞和凋亡[33]。使用CPI-169处理样本可导致多发性骨髓瘤和浆细胞瘤细胞模型的凋亡, 并且能在耐受剂量良好的情况下对异种骨髓瘤模型发挥肿瘤生长抑制作用。而在一组多发性骨髓瘤细胞系中测试EZH2抑制剂和标准治疗(standard of care, SOC)药物的联合研究显示, EZH2抑制剂可以与几种SOC药物在体内外产生协同作用[34]。
CPI-1205在2016年作为高活性小分子EZH2抑制剂被报道(IC50=2 nmol·L-1)。该化合物是由研究人员根据当时已经报道的EZH2抑制剂结构进行一系列基于性质的优化改造得到的(图 4)[35]。以CPI-169为基础, 药物化学工作者对侧链R部分进行了不同类型取代基的尝试。哌啶(N-H)和哌啶(N-Me)衍生物在靶标水平显示出较好的生物学效应, 但是这两个衍生物在细胞水平的生物学机制研究中活性显著下降。将N-H哌啶衍生物改造成脲类、氨基甲酸酯类、磺胺类以及酰胺类化合物可以得到具有生物学活性的类似物, 但是这些类似物在细胞水平的生物学机制研究中仍然没有显示出理想的活性, 同时, N-酰化衍生物还存在体内代谢过快等问题。与N-酰化衍生物相反, 许多由N-H哌啶衍生物改造得到的碱性胺类化合物显示出了理想的药代动力学性质, 为了进一步改进N-H哌啶衍生物的理化性质并得到理想的体内外活性, 研究人员发现取代基碱性的减弱以及由此带来的pKa变化对化合物细胞水平的活性、选择性、毒性以及口服生物利用度等有深远的影响。最终, 合成得到的含三氟甲基的化合物CPI-1205在一系列体内外实验中均表现出了良好的活性与选择性。在Karpas-422异种肿瘤移植模型中, 当给药剂量为160 mg·kg-1 BID时, CPI-1205显示出较强的抗肿瘤活性。CPI-1205结合于PRC2复合物的共晶结构也已经被报道, 这给EZH2抑制剂的后续研发提供了进一步的参考。目前, CPI-1205已经进入临床研究阶段(Phase Ⅰ/Ⅱ), 尝试与单抗药物易普利姆玛(Ipilimumab)联用进行实体肿瘤等的治疗, 也有部分针对进展性B细胞淋巴瘤患者的早期临床研究[36]。值得注意的是, CPI-1205虽然口服利用度高, 但是也存在血浆清除率高的问题。

|
Figure 4 General trends in structure-activity relationships |
CPI-0209是由Constellation Pharmaceuticals研发的最新第二代小分子EZH2抑制剂, 在2019年进入了早期临床试验阶段, 用于晚期实体恶性肿瘤的治疗(NCT04104776), 其具体结构还没有公开。
GSK-126作为一个SAM竞争性的高活性、高选择性EZH2抑制剂[37], 它对EZH2的IC50为9.9 nmol·L-1, 由于其良好的体外试验数据, 曾在葛兰素史克(GlaxoSmithKline, GSK)的Ⅰ期临床试验中用于治疗实体瘤和难治性弥漫性大B细胞淋巴瘤[38]。遗憾的是, 葛兰素史克最终决定终止该产品的开发。作为一个曾经进入临床试验阶段的候选药物, GSK-126口服给药无效, 需要通过静脉滴注给药, 这也是药物化学工作者在进一步研发过程中需要解决的问题之一。
UNC1999是一个通过氢键相互作用以及疏水相互作用结合于EZH2的SAM结合位点的小分子抑制剂(IC50 < 10 nmol·L-1)[39], 其明显特征之一是口服给药有效。由于其血浆浓度在给药后8 h才开始降低, 且能在长达20 h的时间内维持高于细胞水平IC50的浓度, 因此是一个可用于探索EZH2体内功能的工具小分子[40]。
上述现有EZH2吡啶酮类抑制剂具有相似的结构特征(图 5), 通过与SAM竞争性结合于EZH2来发挥作用, 对药物结合位点入口部位残基Y611、Y111以及I109存在获得型突变的EZH2无效[41, 42]。实验证明, 部分肿瘤细胞在EZH2抑制剂的选择性压力下生长时, 会通过特定的突变来获得耐药性, 这也是吡啶酮类抑制剂作为抗肿瘤药物研发的过程中面临的挑战之一[9]。

|
Figure 5 General regions of pyridine inhibitors (Take GSK-126 as an example) |
SHR-2554作为EZH2抑制剂的口服增效剂于2018年进入临床试验(Phase Ⅰ/Ⅱ)阶段, 用于治疗复发或难治性成熟淋巴样肿瘤患者(NCT03603951), 或者与SHR-3680联合用于转移性去势抵抗前列腺癌患者的治疗(NCT03741712)。由江苏恒瑞开发的该候选药物的结构信息尚未公开, 但作为国内首个进入临床试验阶段的EZH2抑制剂, 它的出现表明国内表观遗传相关药物的开发在一定程度上紧跟了国际前沿热点的步伐。
2 靶向EED抑制剂的研究进展EED是PRC2复合物的基本结构单元, PRC2复合物的高分辨率晶体结构表明, EED与H3K27me3的结合导致了EZH2上刺激响应基序(stimulation-responsive motif, SRM)的构象变化, 从而提高了其催化效率[10]。这给了药物化学工作者启发, 可能可以通过开发靶向EED的小分子化合物调节PRC2的活性[43]。
共晶结构显示, EZH2的一段结合于EED上H3K27me3结合位点对面的口袋(图 6)[44], 因此, 靶向EED的小分子抑制剂目前可以分为两大类, 即结合于EED顶部的H3K27me3结合位点的变构抑制剂(图 7)与结合于EED底部的EZH2结合位点的蛋白-蛋白相互作用(protein-protein interaction, PPI)抑制剂(图 8a)。

|
Figure 6 Structural basis of EZH2 recognition by embryonic ectoderm development (EED) |

|
Figure 7 PRC2 complex inhibitors binding to the H3K27me3-binding pocket of EED protein |

|
Figure 8 a: PRC2 complex inhibitors binding to the EBD-binding pocket of EED protein. b: Sequence of SAH-EZH2 |
EED-226是结合于EED顶部H3K27me3结合位点的代表性PRC2抑制剂。在进行PRC2抑制剂的高通量筛选后, 研究人员对得到的非SAM竞争性抑制剂的构效关系(structure-activity relationship, SAR)进行研究, 并开展进一步的药代动力学优化, 最终发现了EED-226[45-47]。它是一个首创(first-in-class)的、选择性、口服有效的EED抑制剂, 当H3K27me0肽用作体外酶法测定的底物时, EED-226抑制PRC2的IC50为23.4 nmol·L-1, 且在所有物种中EED-226均显示出中高水平的血浆蛋白结合能力[48]。在阻断PRC2靶基因H3K27甲基化和诱导人淋巴瘤异种移植瘤消退方面, EED-226显示出了与SAM竞争性抑制剂相似的活性, 值得注意的是, EED-226也能有效抑制包含一种突变的EZH2蛋白的PRC2复合物, 具有该突变的蛋白对SAM竞争性抑制剂存在耐药性。
A-395在2017年被报道为一种强效H3K27me3竞争性EED抑制剂[49]。结构研究表明, A-395结合于EED的H3K27me3结合位点, 从而抑制了PRC2复合物的变构激活(Ki=4 nmol·L-1)。其体内外表型效应与目前已知的PRC2抑制剂相似, 但是A-395能在对其他催化功能抑制剂具有抗性的细胞系中维持有效活性。因此, 它是一种具有代表性的, 能用作机制研究工具的新型PRC2变构抑制剂。
目前, 靶向EED的小分子抑制剂中推进程度最快的是诺华开发的MAK-683, 已经进入临床试验(Phase Ⅰ/Ⅱ)阶段(NCT02900651), 用于治疗成人晚期恶性肿瘤, 如DLBCL、鼻咽癌或其他目前尚无进一步有效标准治疗方法的晚期、复发性或转移性实体肿瘤。
2.2 结合于EED底部的EZH2结合位点的PPI抑制剂研究发现, EZH2的一个30个残基的肽(39-68)对其与EED的相互作用是至关重要的, 该肽与EED复合的晶体结构已被确定(图 6)。结构显示该EED结合域(EED-binding domain, EBD)与EED的WD-重复结构域的底部结合[7]。EED对EBD的具体识别是通过高密度的范德华力和大范围的氢键网络形成的, EBD的C端刚性圈与EED的相互作用由范德华力主导。氢键似乎更有助于EBD的N末端螺旋结合到EED, 围绕该区域形成精细的氢键网络。
有相关报道显示, EZH2经稳定的α螺旋(stabilized alpha-helix of EZH2, SAH-EZH2)可以通过剂量响应性地破坏EZH2-EED复合物和降低EZH2蛋白水平来选择性抑制H3K27me3, 这一机制不同于靶向EZH2催化结构域的小分子[50]。研究人员使用荧光偏振(fluorescence polarization, FP)和免疫共沉淀测定FITC标记的SAH-EZH2肽对EED的结合亲和力, 评估了暴露于FITC标记衍生物的MLL-AF9白血病细胞中SAH-EZH2肽的细胞摄取, 然后对电泳细胞提取物进行了荧光分析。基于EED结合效力和观察到的细胞穿透, SAH-EZH2A (42-68) (图 8b)成为用于优化和生物学测试的先导构建体。SAH-EZH2可以阻止EZH2依赖型白血病细胞的生长和诱导分化, 但是对非致瘤型造血细胞几乎没有明显的影响, 该发现强调了SAH-EZH2的序列和背景依赖型抗增殖活性。
采用生物大分子相互作用分析系统(Biacore 3000)筛选可以结合到EED的天然产物, 并进一步通过免疫共沉淀方法识别PRC2干扰物, 研究人员最终发现了可以结合到EED并且靶向干扰EZH2-EED相互作用的蟛蜞菊内酯(wedelolactone), 它对EED具有较高的亲和力(KD=2.82 μmol·L-1)[51]。在PRC2依赖的肿瘤细胞株中进行了一系列蟛蜞菊内酯对细胞增殖、细胞周期、细胞入侵以及细胞迁移等作用的研究, 结果表明该化合物在PRC2依赖的肿瘤细胞株中可以抑制增殖、诱发凋亡和细胞周期停滞, 并抑制细胞迁移。
2014年, 上海药物所报道了阿司咪唑(Astemizole)作为EZH2-EED PPI抑制剂的发现[8]。该研究首先采用EED-EZH2复合物的晶体结构作为靶标进行基于对接的虚拟筛选, 由于相对“热点”区域集中在EZH2肽的N-末端部分, 最终选取了EZH2 F42 8 Å范围内的残基作为结合位点。筛选结果发现阿司咪唑可竞争性结合于EED, FP实验显示其IC50为93.8 μmol·L-1, Ki值为23.01 μmol·L-1。细胞水平的研究显示, 阿司咪唑可以通过破坏EZH2-EED复合物来削弱PRC2的活性, 并抑制PRC2依赖的淋巴瘤的增殖。
EZH2-EED交界面的可药性已经通过SAH-EZH2肽以及小分子阿司咪唑等得到了证明, 值得注意的是, 这些抑制剂不仅能降低H3K27me3的水平, 还具有更为独特的作用机制, 其中EZH2蛋白质降解过程可在肿瘤细胞中的PRC2复合物解离后被触发[8]。随后的研究表明, SAH-EZH2可以降低H3K27me3和EZH2水平, 并减少具有EZH2酶抑制剂抗药性的SWI-SNF突变肿瘤细胞株的生长。这些研究结果突出了靶向EZH2-EED PPI来治疗EZH2依赖型癌症的应用前景。
然而, EZH2-EED PPI抑制剂仍然存在很多的问题, 肽类抑制剂的代谢稳定性差、生物利用度低, 而阿司咪唑在体外测试中仅显示出中等活性且具有心脏毒性, 这极大阻碍了其在体内治疗方面的应用。
为了找到更多EZH2-EED PPI抑制剂, 药物化学工作者开发并应用了多种新型高通量筛选方法[52], 近期, 报道了一项基于FP的对包含1 600种FDA获批药物的内部化合物库的筛选, 该项筛选初步确定了盐酸阿波吗啡等4种化合物有可能作为潜在的EZH2-EED PPI抑制剂[53]。
3 结语与展望PRC2复合物在多种疾病的发生与进展中发挥作用, 其抑制剂的发现是目前抗肿瘤药物研究领域的热点之一。PRC2的催化活性依赖于其关键催化亚基EZH2, 并且需要在EED及SUZ12的存在下才能发挥其甲基转移酶活性, 因此, 靶向EZH2以及靶向EZH2-EED PPI的小分子抑制剂都可能抑制PRC2的致癌活性。目前, 基于这两种作用机制的小分子抑制剂都已经有了一定的发展。EPZ-6438作为开发进度最快、已经获批进入临床应用的EZH2抑制剂表明了抑制PRC2活性作为相关肿瘤治疗策略的可行性。EZH2-EED PPI抑制剂的开发目前仍处于早期阶段, 通过小分子化合物干扰蛋白-蛋白相互作用是近年来快速发展起来的药物研发新策略, 相关化合物也表现出了全新作用机制, 期待对更多的研究工作及成果的进一步报道。本综述浅述了靶向PRC2相关蛋白小分子抑制剂的研究进展, 为想要了解相关发展动态、开展相关研究的药物化学工作者们提供部分参考, 同时也希望更多的相关候选药物可以进入临床研究或成功上市, 惠及更多患者。
作者贡献: 顾婧是本文的第一作者, 负责文献调研、整理以及文章的撰写; 郭小可为本文的通讯作者, 负责内容框架组织、稿件修改等工作; 尤启冬为本文的共同通讯作者, 负责稿件修改完善等工作。
利益冲突: 本文的研究内容无任何利益冲突。
| [1] |
Laugesen A, Helin K. Chromatin repressive complexes in stem cells, development, and cancer[J]. Cell Stem Cell, 2014, 14: 735-751. DOI:10.1016/j.stem.2014.05.006 |
| [2] |
Cao R, Wang L, Wang H, et al. Role of histone H3 Lysine 27 methylation in polycomb-group silencing[J]. Science, 2002, 298: 1039-1043. DOI:10.1126/science.1076997 |
| [3] |
Jiao LY, Liu X. Response to comment on "structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2"[J]. Science, 2016, 354: 1543-1543. |
| [4] |
Portoso M, Ragazzini R, Rrencic Z, et al. PRC2 is dispensable for HOTAIR-mediated transcriptional repression[J]. EMBO J, 2017, 36: 981-994. DOI:10.15252/embj.201695335 |
| [5] |
Beltran M, Yates CM, Skalska L, et al. The interaction of PRC2 with RNA or chromatin is mutually antagonistic[J]. Genome Res, 2016, 26: 896-907. DOI:10.1101/gr.197632.115 |
| [6] |
Czermin B, Melfi R, Mccabe D, et al. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal polycomb sites[J]. Cell, 2002, 111: 185-196. DOI:10.1016/S0092-8674(02)00975-3 |
| [7] |
Han Z, Xing X, Hu M, et al. Structural basis of EZH2 recognition by EED[J]. Structure (Cambridge), 2007, 15: 1306-1315. DOI:10.1016/j.str.2007.08.007 |
| [8] |
Kong X, Chen L, Jiao L, et al. Astemizole arrests the proliferation of cancer cells by disrupting the EZH2-EED interaction of polycomb repressive complex 2[J]. J Med Chem, 2014, 57: 9512-9521. DOI:10.1021/jm501230c |
| [9] |
Brooun A, Gajiwala KS, Deng YL, et al. Polycomb repressive complex 2 structure with inhibitor reveals a mechanism of activation and drug resistance[J]. Nat Commun, 2016, 7: 11384. DOI:10.1038/ncomms11384 |
| [10] |
Justin N, Zhang Y, Tarricone C, et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2[J]. Nat Commun, 2016, 7: 11316. DOI:10.1038/ncomms11316 |
| [11] |
Kouznetsova VL, TchekanovA, Li XM, et al. Polycomb repressive 2 complex-molecular mechanisms of function[J]. Protein Sci, 2019, 28: 1387-1399. DOI:10.1002/pro.3647 |
| [12] |
Gorodetska I, Lukiyanchuk V, Peitzsch C, et al. BRCA1 and EZH2 cooperate in regulation of prostate cancer stem cell phenotype[J]. Int J Cancer, 2019, 145: 2974-2985. DOI:10.1002/ijc.32323 |
| [13] |
Sabour TJ, Aghaie FA, Mohammidi S, et al. Indirect tumor inhibitory effects of microRNA-124 through targeting EZH2 in the multiple myeloma cell line[J]. Cell J, 2020, 22: 23-29. |
| [14] |
Ohuchi M, Sakamoto Y, Tokunaga R, et al. Increased EZH2 expression during the adenoma carcinoma sequence in colorectal cancer[J]. Oncol Lett, 2018, 16: 5275-5281. |
| [15] |
Zingg D, Arenas-Ramirez N, Sahin D, et al. The histone methyltransferase EZH2 controls mechanisms of adaptive resistance to tumor immunotherapy[J]. Cell Rep, 2017, 20: 854-867. DOI:10.1016/j.celrep.2017.07.007 |
| [16] |
Comet I, Riising EM, Leblanc B, et al. Maintaining cell identity:PRC2-mediated regulation of transcription and cancer[J]. Nat Rev Cancer, 2016, 16: 803-810. DOI:10.1038/nrc.2016.83 |
| [17] |
Cholewa-Waclaw J, Bird A, Von Schimmelmann M, et al. The role of epigenetic mechanisms in the regulation of gene expression in the nervous system[J]. J Neurosci, 2016, 36: 11427-11434. DOI:10.1523/JNEUROSCI.2492-16.2016 |
| [18] |
Hirabayashi Y, Suzki N, Tsuboi M, et al. Polycomblimits the neurogenic competence of neural precursor cells to promote astrogenic fate transition[J]. Neuron, 2009, 63: 600-613. DOI:10.1016/j.neuron.2009.08.021 |
| [19] |
Pereira JD, Sansom SN, Smith J, et al. EZH2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex[J]. Proc Natl Acad Sci U S A, 2010, 107: 15957-15962. DOI:10.1073/pnas.1002530107 |
| [20] |
Jung M, Pfeifer GP. Aging and DNA methylation[J]. BMC Biol, 2015, 13: 7. DOI:10.1186/s12915-015-0118-4 |
| [21] |
Xun H, Juan Y, Min Z, et al. Targeting epigenetic crosstalk as a therapeutic strategy for EZH2-aberrant solid tumors[J]. Cell, 2018, 175: 1-14. DOI:10.1016/j.cell.2018.08.026 |
| [22] |
Tan J, Yang X, Zhuang L, et al. Pharmacologic disruption of polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells[J]. Gen Devel, 2007, 21: 1050-1063. DOI:10.1101/gad.1524107 |
| [23] |
El Ouardi D, Mouhamed I, Anna S, et al. The inhibition of the histone methyltransferase EZH2 by DZNEP or siRNA demonstrates its involvement in MGMT, TRA2A, RPS6KA2, and U2AF1 gene regulation in prostate cancer[J]. OMICS, 2019, 24: 116-118. |
| [24] |
Kikuchi J, Takashina T, Kinoshita I, et al. Epigenetic therapy with 3-deazaneplanocin A, an inhibitor of the histone methyltransferase EZH2, inhibits growth of non-small cell lung cancer cells[J]. Lung Cancer, 2012, 78: 138-143. DOI:10.1016/j.lungcan.2012.08.003 |
| [25] |
Sun F, Lee L, Zhang Z, et al. Preclinical pharmacokinetic studies of 3-deazaneplanocin A, a potent epigenetic anticancer agent, and its human pharmacokinetic prediction using GastroPlus?[J]. Eur J Pharm Sci, 2015, 77: 290-302. DOI:10.1016/j.ejps.2015.06.021 |
| [26] |
Knutson SK, Wigle TJ, Warholic NM, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells[J]. Nat Chem Biol, 2012, 8: 801-816. |
| [27] |
Lindsay CD, Kostiuk MA, Harris J, et al. Efficacy of EZH2 inhibitory drugs in human papillomavirus-positive and human papillomavirus-negative oropharyngeal squamous cell carcinomas[J]. Clin Epigenet, 2017, 9: 95. DOI:10.1186/s13148-017-0390-y |
| [28] |
Kuntz K, Keilhack H, Pollock R, et al. The discovery and pre-clinical development of the first clinical stage EZH2-inhibitor, EPZ-6438(E7438)[J]. Eur J Cancer, 2014, 50: 92. |
| [29] |
Knutson SK, Kawano S, Minoshima Y, et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma[J]. Mol Cancer Ther, 2014, 13: 842-854. DOI:10.1158/1535-7163.MCT-13-0773 |
| [30] |
Brach D, Johnston-Blackwell D, Drew A, et al. EZH2 inhibition by tazemetostat results in altered dependency on B-cell activation signaling in DLBCL[J]. Mol Cancer Ther, 2017, 16: 2586-2597. DOI:10.1158/1535-7163.MCT-16-0840 |
| [31] |
Chen J, Chen X, Yao J, et al. The combination of decitabine and EPZ-6438 effectively facilitate adipogenic differentiation of induced pluripotent stem cell-derived mesenchymal stem cells[J]. Biochem Biophys Res Commun, 2019, 516: 307-312. DOI:10.1016/j.bbrc.2019.06.093 |
| [32] |
Campbell JE, Kuntz KW, Knutson SK, et al. EPZ011989, a potent, orally-available EZH2 inhibitor with robust in vivo activity[J]. ACS Med Chem Lett, 2015, 6: 491-495. DOI:10.1021/acsmedchemlett.5b00037 |
| [33] |
Balasubramanian V, Iyer P, Arora S, et al. Abstract 1697:CPI-169, a novel and potent EZH2 inhibitor, synergizes with CHOP in vivo and achieves complete regression in lymphoma xenograft models[J]. Cancer Res, 2014, 74: 19. |
| [34] |
Shilpi A, Kaylyn W, Shruti A, et al. EZH2 inhibitors are broadly efficacious in multiple myeloma as single agent and in combination with standard of care therapeutics[J]. Blood, 2016, 128: 5672. DOI:10.1182/blood.V128.22.5672.5672 |
| [35] |
Vaswani RG, Gehling VS, Dakin LA, et al. Identification of (R)-N-((4-methoxy-6-methyl-2-oxo-1, 2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2, 2, 2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a potent and selective inhibitor of histone methyltransferase EZH2, suitable for Phase Ⅰ Clinical Trials for B-Cell lymphomas[J]. J Med Chem, 2016, 59: 9928-9941. DOI:10.1021/acs.jmedchem.6b01315 |
| [36] |
Taplin ME, Hussain A, Shah S, et al. ProSTAR:a Phase Ib/Ⅱ study of CPI-1205, a small molecule inhibitor of EZH2, combined with enzalutamide (E) or abiraterone/prednisone (A/P) in patients with metastatic castration-resistant prostate cancer (mCRPC)[J]. J Clin Oncol, 2019, 37: 7. |
| [37] |
McCabe MT, Ott HM, Ganji G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations[J]. Nature, 2012, 492: 108-112. DOI:10.1038/nature11606 |
| [38] |
Zeng D, Liu M, Pan J. Blocking EZH2 methylation transferase activity by GSK126 decreases stem cell-like myeloma cells[J]. Oncotarget, 2016, 8: 3396. |
| [39] |
Konze KD, Ma A, Li F, et al. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1[J]. ACS Chem Biol, 2013, 8: 1324. DOI:10.1021/cb400133j |
| [40] |
Liang J, Li Q, Cai W, et al. Inhibition of polycomb repressor complex 2 ameliorates neointimal hyperplasia by suppressing trimethylation of H3K27 in vascular smooth muscle cells[J]. Br J Pharmacol, 2019, 176: 3206-3219. |
| [41] |
Lue JK, Amengual JE. Emerging EZH2 inhibitors and their application in lymphoma[J]. Curr Hematol Malign Rep, 2018, 13: 369-382. DOI:10.1007/s11899-018-0466-6 |
| [42] |
Matthew B, Xin Y, Xin L. An evolutionarily conserved structural platform for PRC2 inhibition by a class of EZH2 inhibitors[J]. Sci Rep, 2018, 8: 9092. DOI:10.1038/s41598-018-27175-w |
| [43] |
Lingel A, Sendzik M, Huang Y, et al. Structure-guided design of EED binders allosterically inhibiting the epigenetic polycomb repressive complex 2(PRC2) methyltransferase[J]. J Med Chem, 2017, 60: 415-427. DOI:10.1021/acs.jmedchem.6b01473 |
| [44] |
Yang CY, Wang S. Allosteric inactivation of polycomb repressive complex 2(PRC2) by inhibiting its adapter protein:embryonic ectodomain development (EED)[J]. J Med Chem, 2017, 60: 2212-2214. DOI:10.1021/acs.jmedchem.7b00287 |
| [45] |
Qi W, Zhao K, Gu J, et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED[J]. Nat Chem Biol, 2017, 13: 381-388. DOI:10.1038/nchembio.2304 |
| [46] |
Huang Y, Zhang J, Yu Z, et al. Discovery of first-in-class, potent and orally bioavailable EED inhibitor with robust anti-cancer efficacy[J]. J Med Chem, 2017, 60: 2215-2226. DOI:10.1021/acs.jmedchem.6b01576 |
| [47] |
Li L, Zhang HL, Zhang M, et al. Discovery and molecular basis of a diverse set of polycomb repressive complex 2 inhibitors recognition by EED[J]. PLoS One, 2017, 12: e0169855. DOI:10.1371/journal.pone.0169855 |
| [48] |
Zhang JY. Preclinical pharmacokinetics and metabolism of a first-in-class, potent and orally bioavailable EED inhibitor EED226[J]. Drug Metab Pharmacokinet, 2018, 33: S42. |
| [49] |
He Y, Selvaraju S, Curtin ML, et al. The EED protein-protein interaction inhibitor A-395 inactivates the PRC2 complex[J]. Nat Chem Biol, 2017, 13: 389. DOI:10.1038/nchembio.2306 |
| [50] |
Kim W, Bird GH, Neff T, et al. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer[J]. Nat Chem Biol, 2013, 9: 643-650. DOI:10.1038/nchembio.1331 |
| [51] |
Chen HM, Guo SJ, Li JD, et al. Wedelolactone disrupts the interaction of EZH2-EED complex and inhibits PRC2-dependent cancer[J]. Oncotarget, 2015, 6: 13049-14059. DOI:10.18632/oncotarget.3790 |
| [52] |
Jon AR, Jonathan T, Philip BR, et al. Rapid identification of novel allosteric PRC2 inhibitors[J]. ACS Chem Biol, 2019, 14: 2134-2140. |
| [53] |
Zhu MR, Du DH, Hu JC, et al. Development of a high-throughput fluorescence polarization assay for the discovery of EZH2-EED interaction inhibitors[J]. Acta Pharmacol Sin, 2018, 39: 302-310. DOI:10.1038/aps.2017.59 |