 2020, Vol. 55
2020, Vol. 55

新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多维性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
20世纪50年代氯丙嗪问世, 开创了药物治疗精神失常的新领域。三环类药物作为经典抗精神病药物, 存在锥体外症候等不良反应。阿立哌唑是致力于消除不良反应的非经典抗精神失常药物研究成功范例之一。基于对突触前多巴胺自受体和突触后多巴胺受体的生理功能的认识, 大冢制药的研究者设定研发作用于双环节 (而非双靶标) 的新型抗精神失常药物的目标, 采用实验动物的生理表型变化评价化合物活性与不良反应, 以传统药物化学和构效关系分析方法设计合成的化合物, 将药效、药代和安全性在整体动物的“综合平台”上同步优化。后继的研究证明, 阿立哌唑是多靶标作用的药物, 对D2、D3和5-HT受体都有较高的亲和力, 尤其是5-HT2A与D2亲和力的高比值, 是消除不良反应的必要条件。基于生理表型的药物研究与基于靶标的分子设计是互为关联和异途同归的策略, 阿立哌唑提供了一个佐证。
(编者按)
精神分裂症公认的发病机制是多巴胺学说, 认为是患者脑内多巴胺(DA)神经元系统的过度活化所致, 因而研发抗精神病药物的策略是抑制突触后的多巴胺受体, 阻断DA的神经传递。临床上应用的抗精神病药物都是通过阻断突触后DA受体抑制DA的神经传导。然而单纯的DA受体拮抗剂可引起许多不良反应, 例如锥体外综合征、帕金森病样症状, 以及长时间抑制DA受体导致脑中DA受体超敏性引起的迟发性运动障碍(不随意运动的症状)等。消除不良反应可通过刺激突触前DA自受体以降低多巴胺样的神经传导。
1.2 多巴胺自受体的功能和部分激动剂的作用多巴胺自受体(DA autoreceptor)是位于神经末梢突触前膜的DA受体, DA刺激DA自受体会反馈性地对DA的释放产生负性或正性调节作用。多数情况下, 激活自受体所引发的作用是抑制性的, 即DA一旦释放到神经元附近的细胞外液中, 导致DA的释放与合成频率降低。所以DA自受体是调控DA释放量的一个环节, DA释放量过多时, 自受体抑制DA的合成和释放; 释放不足时, 自受体促进合成和释放。因此, 如果DA受体的部分激动剂对自受体产生激动作用, 从而减少多巴胺的过度活跃, 同时又对突触后DA受体有弱拮抗作用, 可成为较少不良反应的抗精神病药物。
2 先导物及其优化 2.1 活性评价在研发阿立哌唑的20世纪80年代, 还没有评价DA受体的离体实验模型, 活性(和不良反应)的评价都是以动物的表型变化(测定小鼠行为指标)评价化合物的作用。所以, 这个项目是用动物的多个宏观指标反映对不同受体的微观调控作用来评价化合物的活性和不良反应。①抑制小鼠跳跃实验:活性越高越好。多巴胺引起小鼠不停地跳跃, 化合物的神经安定作用与抑制小鼠跳跃的活性相关。方法是灌胃不同剂量的受试物, 45 min后腹腔注射甲基苯丙胺(抑制多巴胺的释放)和多巴(在体内代谢成多巴胺), 造成小鼠跳跃的模型。注射10 min后, 计数小鼠在50 min内跳跃的次数, 计算跳跃次数低于10次的半数有效剂量(ED50, mg·kg-1)作为受试物对突触前DA的释放和合成的抑制活性, 即对DA自受体的激动作用。②诱导小鼠僵住实验:这是评价受试物对锥体外的不良反应, 以没有或低活性为佳。方法是受试物灌胃小鼠后, 在0、1、2、4、6和8 h将小鼠前肢摆成非自然状态, 若维持30 s以上, 而且连续3次都维持30 s以上, 即判为僵住。计算引起50%动物发生僵住的剂量(ED50, mg·kg-1)。③ α肾上腺能受体阻断作用:是评价受试物的另一种不良反应, 以没有或低阻断活性为佳。方法是小鼠灌胃受试物, 60 min后腹腔注射去甲肾上腺素, 观察24 h动物死亡数, 动物存活表明具有α肾上腺能受体阻断作用, 计算ED50 (mg·kg-1)。
由于受试物是灌胃给药, 可以判断化合物的吸收状况, 虽然不是精确和定量的评价药代动力学性质, 但从药效-剂量的关系上反映了胃肠道吸收和穿越血脑屏障的能力。此外, 除上述僵住实验和α肾上腺能受体阻断实验外, 动物的一般行为表现也在一定程度上反映了药物的毒副作用。
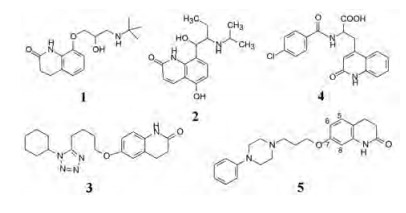
2.2 先导化合物及其优化 2.2.1 先导化合物大冢制药的药物化学家在20世纪80年代(甚至更早)研发新药的化学结构, 常以喹啉酮为母核, 例如1980年上市的非选择性β肾上腺能阻断剂卡替洛尔(1, caeteolol)、1981年上市的β肾上腺能激动剂丙卡特罗(2, rocaterol)、1988年上市的磷酸二酯酶(PDE) III抑制剂西洛他唑(3, cilostazole)以及胃黏膜保护剂瑞巴派特(4, rebanipide)等, 都以(二氢)喹啉酮为结构骨架。
在研发无镇静作用的抗过敏药物的过程中, 发现含有喹啉酮结构的化合物5具有抗精神失常样活性, 可抑制甲基苯丙胺引起小鼠的跳跃行为, 也不发生小鼠的锥体外不良反应(如僵住状态)。化合物5被认作先导化合物。
|
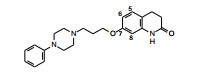
首先考察苯基哌嗪侧链连接在喹唑啉环上的不同位置对活性和安全性的影响, 设计合成的有代表性的化合物列于表 1。结果显示, 变换侧链的位置可影响化合物的活性和安全性。化合物6的侧链连接在5位, 与先导物5相比, 提高了抑制小鼠跳跃活性, 但也提高了阻断α肾上腺能作用, 活性与不良反应相近。连接在6位的化合物7, 对去甲肾上腺素的拮抗作用尤其显著, 不良反应超过了活性。化合物8未显示活性。因而不拟在6和8位作进一步优化。
| Table 1 The effect of side chains at different position on the activity and safety |
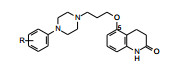
考察连接在母核5位的各种取代的苯基哌嗪对活性的影响, 有代表性的化合物列于表 2。结果提示, 苯环引入不同取代基都使活性提高, 但抗α肾上腺能的不良反应也增强, 苯环取代使活性和不良反应并进, 因而不宜研发5位连接的侧链。
| Table 2 Structures and activity of 5-substituent of phenyl compounds |
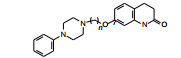
在变换苯环上取代基之前, 首先考察侧链的亚烷基长度对活性的影响, 表 3的化合物活性提示碳原子数与抑制小鼠跳跃作用的关系是: n = 3 (化合物5) ≧ n = 4 (化合物13) > > n = 2 (化合物12)和n = 5 (化合物14), 因而进一步优化结构固定为7位的三亚甲基链。
| Table 3 The effect of chain length on the activity |
在苯环上作了密集的取代基变换, 包括不同位置的单或二取代化合物列于表 4。分析化合物的构效关系, 提示苯环上引入一个或两个取代基可提高抑制甲基苯丙胺引起的小鼠跳跃活性, 但2, 3-二氯化合物(25)的活性显著下降, 是个例外。一些化合物的活性强于氟哌啶醇, 例如2-CH3 (22)、3-CH3 (23)、2, 3-(CH3)2 (31)等, 活性与氟哌啶醇相当的化合物如2-F (17)和4-CH3 (24)。苯环引入取代基也提高了抑制α肾上腺能作用, 而且单取代的作用强于二取代化合物。化合物诱导僵住作用(也是不良反应)与抑制甲基苯丙胺的跳跃活性不呈相关性, 一些高活性的化合物诱导僵住作用较弱。31是活性高和选择性强的化合物。
| Table 4 The effect of substituted phenyl compounds on the activity |
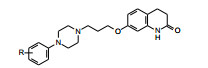
以上合成的化合物都是以二氢喹啉酮为母核。对有代表性的化合物合成了含有双键的喹啉酮类似物列于表 5。总的趋势是二氢喹啉酮的抑制小鼠跳跃活性略强于相应的喹啉酮化合物, 对不良反应的影响没有显示出相关性。
| Table 5 Effect of saturation of quinolinone on the activity |
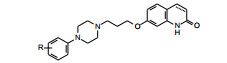
抗精神分裂药物如氯丙嗪和氟哌啶醇由于阻断了DA受体, 经负反馈机制增加DA的释放量, 使得大鼠脑中提高了DA代谢产物二羟基苯乙酸(DOPAC)的水平, 而脑中DOPAC浓度提高是不良反应的指证。对上述高活性化合物评价了氟哌啶醇引起DOPAC水平提高的抑制作用, 列于表 6。
| Table 6 Effects of 2(1H)-quinolinone derivatives on the increase in concentration of 3, 4-dihydroxphenylacetic acid (DOPAC) induced by haloperidol in discrete brain region of the rat |
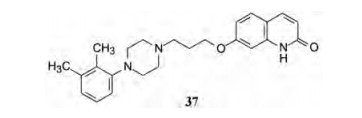
化合物16、23和37可以阻断氟哌啶醇诱导提高大鼠脑各个部位的DOPAC浓度, 而且可阻断甲基苯丙胺对小鼠的毒性, 提示这3个化合物对于DA自受体具有激动作用。对化合物16 (代号OPC-4139)和37(OPC-4392)作深入研究, 显示37对突触前多巴胺自受体有激动作用和对突触后多巴胺D2受体有拮抗作用(Yasuda Y, Kikuchi T, Suzuki S, et al. 7-[3-[4-(2, 3-Dimethylphenyl)-piperazinyl]propoxy]-2(1H)-quinolinone (OPC-4392), a presynaptic dopamine autoreceptor agonist and postsynaptic D2-receptor antagonist. Life Sci, 1988, 42: 1941-1954), 确定37进入临床实验(Banno K, Fujioka T, Kikuchi T, et al. Studies on 2(1H)-quinolinone derivatives as neuroleptic agents, I. Synthesis and biological activities of (4-phenyl-1-piperazinyl)-propoxy-2(1H)-quinolinone derivatives. Chem Pharm Bull, 1988, 36: 4377-4388)。
临床研究表明37作为突触前多巴胺自受体的激动剂对于治疗精神分裂症的负性症状是有效的, 作为突触后的多巴胺受体的拮抗剂也可治疗患者的正性症状。
|
化合物37在进行临床研究的同时, 大冢制药还以37为起始物, 继续优化结构。
3.1 新一轮评价活性的模型① 评价化合物对突触后多巴胺受体的拮抗作用。用多巴胺受体激动剂阿扑吗啡造模进行小鼠整体实验。方法是灌胃受试物后皮下注射一定量阿扑吗啡, 引起小鼠的刻板状态, 在一定的时间内评价受试化合物抑制半数动物刻板行为的剂量(ED50, μmol·kg-1)。②评价化合物对突触前多巴胺自受体的激动作用。方法是受试物灌胃小鼠, 用γ-丁内酯(GBL)引起小鼠脑内合成多巴胺增高, 测定50%小鼠达到指定水平的化合物剂量(ED50, μmol·kg-1)。③评价化合物诱导僵住作用, 方法同前述, 用ED50 (μmol·kg-1)表示。④评价化合物对α肾上腺能的拮抗作用。方法同前述, 用ED50 (μmol·kg-1)表示。
3.2 优化过程和构效关系化合物37在进行临床研究的同时, 继续作结构优化。用小鼠阿扑吗啡模型评价37对DA受体的拮抗活性ED50为41.3 μmol·kg-1。将37的喹啉酮还原成二氢化合物38, 提高了活性。将苯环上的2, 3-二甲基换成2-甲基-3-氯化合物39, 活性提高, 提示亲脂性和(或)拉电子性都有利于活性。
根据前述的构效关系经验, 侧链的碳原子3~4个为优选长度, 二氢喹啉酮母核是否仍然如此, 因而合成了四碳和五碳相连的化合物40和41, 表明40的活性强于39, 而41无活性。因而以化合物40为新一轮的先导化合物。表 7列出了化合物37~41的结构与活性。
| Table 7 Structure evolution of lead in a new turn. * ED50 > 23 μmol·kg-1 represents no activity |
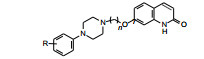
以化合物40作为新的起始物, 在苯环的不同位置连接不同数目和性质的基团, 合成的化合物列于表 8。活性评价指标是对DA受体的拮抗作用。构效关系分析如下: ①保持2-甲基不变, 变换3位的氯成溴原子(拉电子性相同, 脂溶性提高, 体积加大)化合物43仍保持活性; 换成体积较小的氟原子(45)活性减弱; 拉电子更强的硝基化合物48活性显著提高, 而氰基(47)活性略降。推电子基团氨基(49)、乙酰氨基(50)和羟基(51)失去活性。结论:拉电子和体积较大基团有利于活性。②起始物40的甲基与氯的位置互换, 化合物52的活性提高, 同样将43的甲基与溴互换位置, 得到的53活性也提高, 提示2位的拉电子基团有利于活性, 因而又合成了2, 3-二氯化合物55, 活性显著提高。变换氯原子的位置或换成二溴或三氯化合物都使活性减弱。提示2, 3-二氯化合物是优良的取代基。③单取代的卤素取代的化合物活性弱于双取代, 取代位置仍以2位活性强于其他位置。强拉电子的硝基(73)或氰基(75)单取代化合物未显示活性, 而2-甲基(70)、2-甲氧基(71)和2-乙氧基(72)的活性强, 但2-氨基(74)没有活性。提示单取代的构效关系与二取代化合物有很大不同, 说明涵盖了药效与药代的整体动物的表型变化与化学结构关系的复杂性。
| Table 8 Optimization of substituted phenyl ring |
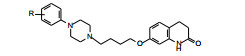
虽然第一轮的优化已经证明了侧链与喹唑酮环连接的位置以7位占优, 但将连接基变换为丁氧基后是否构效关系有变, 因而以高活性的55为参照物, 合成了二氢喹啉酮在5-、6-和8-位连接侧链的化合物(76~78), 列于表 9中。结果表明与前述的结论相同, 侧链未与7位相连的化合物都失去活性。
| Table 9 Activity of compounds with side chain at different position |
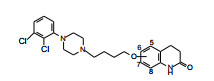
新的链长和苯环上的不同取代对于二氢喹啉酮母核的适配性进行了再考察。将表 7中高活性化合物的二氢喹啉酮换成喹啉酮环, 合成的化合物79~83列于表 10。结果表明, 79~83与相应的二氢喹啉酮化合物40、43、54、55和73相近。
| Table 10 The effect of substituted quinolinone rings on the activity |
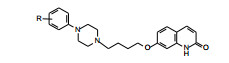
本项目的目标是①阻断突触后DA受体(表 10中DA拮抗); ②激动突触前DA自受体(自受体); ③没有或较低的僵住作用(僵住); ④没有或较低阻断α肾上腺能作用(α受体)。下面对DA受体有较高抑制活性的化合物用其他模型作药理评价。前两项活性越高越好(ED50值小), 后两项越弱越好(ED50值大)。引起僵住作用与DA受体的拮抗作用的比值表示安全范围, 数值越大越安全。表 11列出了这些化合物的药理作用。
| Table 11 Multi-pharmacological activity of compounds. *Cat/DA ant ratio: Catalepsy/DA antagonism ratio |
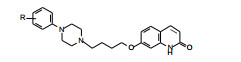
化合物40、54、55、79和82是较强DA受体拮抗剂和DA自受体激动剂, 对α肾上腺能受体呈现弱拮抗作用, 然而化合物54和82 (苯基上有2, 3-二甲基取代)灌胃30 mg·kg-1阿扑吗啡在6 h内不能抑制刻板行为, 而40和79 (苯基上有2-甲基-3-氯取代)则可抑制刻板作用。化合物55 (苯基上有2, 3-二氯取代)活性更强(Oshiro Y, Sato S, Kurahashi N, et al. Novel antipsychotic agents with dopamine autoreceptor agonist properties: synthesis and pharmacology of 7-[4-(4-phenyl-1-piperazinyl)butoxy]-3, 4-dihydro-2(1H)-quinolinone derivatives. J Med Chem, 1998, 26: 658-667)。
5 确定候选化合物和阿立哌唑的上市化合物55在灌胃2 h后可完全抑制刻板状态(抑制阿扑吗啡作用), ED50=11.8 mg·kg-1。对γ丁内酯诱导大鼠脑中多巴胺合成的抑制作用, ED50为6.9 mg·kg-1, 灌胃引起大鼠僵住状态的ED50=149.2 mg·kg-1, 是抑制阿扑吗啡引起小鼠刻板状态剂量的10倍。55的多靶标作用和较低不良反应, 优于已经进入临床研究的化合物37, 确定55为新的候选化合物, 定名阿立哌唑(aripiprazole, OPC-14597), 大冢与美国BMS公司合作进行临床研究。阿立哌唑体内动力学呈现线LIPZ性过程, 口服生物利用度为90%, 给药后3~5 h血浆达到Cmax, 消除半衰期为75 h, 肝脏CYP3A4和2D6是阿立哌唑发生脱氢、羟化和N-去烷基化等的代谢酶。连续用药者脑中阿立哌唑浓度增加, 10~14天达到恒定水平。经Ⅲ期临床研究证明是具有较少不良反应的抗精神病药, 于2002年经FDA批准上市。
|
基于对脑神经突触前DA自受体的生理功能和突触后DA受体的过度兴奋而引发的精神病的认识, 从20世纪80年代开始基于生理表型而研发的新一代抗精神病药物阿立哌唑, 通过药物化学和构效关系的研究, 确保了研制的成功。虽然当时对各种神经递质的受体及其亚型尚不清楚, 更谈不上基于受体结构的分子设计, 但后继的研究表明, 阿立哌唑的安全有效性是与作用于多种受体密切相关的, 例如有高度结合作用的受体有多巴胺D2 (Ki=1.6 nmol·L-1, 部分激动剂)、D3 (Ki=5.4 nmol·L-1, 部分激动剂)、5HTIA (Ki=5.6 nmol·L-1, 部分激动剂)和5HT2A (Ki=8.7 nmol·L-1)、5HT2B (Ki=0.36 nmol·L-1), 中度亲和力的受体有多巴胺D4、5HT2C、5-HT7和H1受体等。现已清楚, 多靶标作用是治疗神经精神病药物的必不可少的因素。阿立哌唑的研制揭示了宏观的表型研究与微观的分子靶标作用是关联的。