 2020, Vol. 55
2020, Vol. 55



2. 中国医学科学院、北京协和医学院药物研究所, 北京 100050;
3. 菏泽市立医院, 山东 菏泽 274000
2. Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, China;
3. Heze Municipal Hospital, Heze 274000, China
肿瘤免疫治疗是以激发和增强患者自身免疫系统为目的, 借助机体免疫系统来杀死肿瘤细胞。肿瘤免疫治疗是目前最有希望能成功治愈癌症的手段, 已成为肿瘤治疗领域中最具前景的研究方向之一。
多种肿瘤的发病都与色氨酸代谢异常有关, 吲哚胺-2, 3-双加氧酶(IDO)是体内催化色氨酸沿犬尿酸途径代谢的限速酶, 可使色氨酸裂解为犬尿酸, 目前研究较多的是IDO1[1, 2]。IDO1在肿瘤组织中高表达, 导致色氨酸的消耗和犬尿酸的形成, 引起T淋巴细胞的增殖抑制, 从而导致免疫抑制, 使肿瘤细胞免受免疫细胞的攻击, 即发生免疫逃逸[3, 4]。通过IDO1抑制, 能够有效调节色氨酸的代谢、促进T细胞增殖、使免疫系统识别和清除肿瘤细胞的功能恢复, 进而攻击和杀死肿瘤细胞, IDO1被认为是肿瘤免疫治疗药物开发的重要靶点[5, 6]。
尤其近几年, IDO1抑制剂的研究热度达到了顶峰, 数十个活性化合物处于临床前研究阶段, 几个进入了临床研究, 其中最快的epacadostat已进入Ⅲ期临床。但去年初, 随着epacadostat Ⅲ期临床研究的失败[7], IDO1抑制剂的研究热度降了下来, 人们开始反思在临床上为什么单独IDO1酶抑制剂疗效甚微, 与PD-L1单抗联合用药也未能达到预期效果。其实, 芝加哥西北大学范伯格医学院Wainwright教授研究小组[8]2012年就报道, 在小鼠原位脑瘤模型中, 肿瘤细胞中的IDO1不是非肿瘤细胞的IDO1, 有助于增加调节性T细胞的积累, 降低小鼠生存期。2014年又有报道[9], 非肿瘤细胞的IDO1的活性有助于免疫检查点抑制导致的小鼠最大生存效益。这表明肿瘤细胞的IDO1和非肿瘤细胞的IDO1对肿瘤免疫功能的影响可能不同。因此, 选择性抑制肿瘤组织内的IDO1, 而不抑制非肿瘤组织内IDO1和机体其他组织内IDO1的抗肿瘤效果可能也不同。为验证这一假设, 本文设计合成了特异性靶向肿瘤组织的IDO1抑制剂前药。
要设计合成特异性靶向肿瘤组织的IDO1抑制剂前药, 首先要了解不同于机体正常组织的肿瘤组织的特征。已知缺氧是大多数肿瘤的特征, 肿瘤缺氧导致了对化疗与放射治疗的抗性与耐药性以及与之相联的癌症预后差、异常血管的生成以及肿瘤脉管系统形成等[10]。并且在低氧条件下, 观察到细胞内还原酶的过表达, 包括硝基还原酶、DT-心肌黄酶和偶氮还原酶等, 在实体瘤中, 硝基还原酶的表达与缺氧状态直接相关[11, 12]。肿瘤微环境缺氧的重要特征正在被用来实现特异性靶向肿瘤组织的治疗。
另外要考虑的一个重要问题就是原药的选择, 化合物B (IC50: 59 nmol·L-1)与epacadostat (IC50: 73 nmol·L-1)对IDO1的抑制活性IC50值都在纳摩尔级别, 但化合物B其理化性质及生物利用度不如epacadostat, 未进入临床研究, 而epacadostat研究的比较多。在临床前研究化合物B与epacadostat的代谢时, 发现它们半衰期较短, 代谢快, 并检测鉴定了化合物B有两种主要的代谢产物(路线图 1), epacadostat有3种主要的代谢产物(路线图2), 这些代谢产物都对IDO1无抑制活性[13]。其结构中肟羟基是一个最重要的活性基团, 也是一个主要代谢位点, 所以选择化合物B和epacadostat作为原药, 基于肟羟基设计前药应该是不错的选择。

|
Figure 1 Structural formula of the designed prodrug compound |

|
Scheme 1 Two metabolites of compound B |
本研究的前药设计思路是在原药化合物B和epacadostat分子的主要代谢位点羟基上键合一个能够在肿瘤缺氧的微环境中降解的载体分子, 这个键合过程既要求在实验室能简便完成和高效制备, 在体内肿瘤组织中又可以完全的逆转释放出原药, 而且前药在体内到达肿瘤组织之前要相对稳定和低毒性。本文选择已报道的硝基芳环化合物, 如硝基取代苄基、硝基取代呋喃甲基、硝基取代吡咯甲基等作为载体分子, 与化合物B和epacadostat分子的羟基偶联形成能够特异性靶向肿瘤组织的前药(图 1)。因为在肿瘤缺氧微环境中, 载体分子上的硝基能够被高表达的硝基还原酶等还原为胺中间体, 由于共轭效应此中间体不稳定发生断裂, 释放IDO1抑制剂(路线图3)。

|
Scheme 2 Three metabolites of epacadostat |
总之, 基于肿瘤微环境缺氧的重要特征, 以研究最多和发展最快的羟基脒类IDO1抑制剂, 如化合物B和epacadostat为原药, 并通过封堵化合物B和epacadostat的主要代谢位点, 设计合成特异性靶向肿瘤组织的, 并能够提高代谢稳定性、降低不良反应的新型IDO1抑制剂前药。
本文所设计的前药化合物合成分为三部分, 首先是原药化合物B与epacadostat的合成; 然后载体分子溴代物的合成; 最后是二者的键合连接。前面两部分是按照文献进行合成[14, 15], 而最后部分原药与载体分子的连接则是采用碱性环境下原药肟羟基与载体分子溴代物的亲核取代反应(路线图4)。
结果与讨论 1 化合物的合成经过实验探索发现:在合成路线4的a步中选用碳酸钾为碱, DMF为溶剂, 反应效果较好。此步反应曾尝试使用碳酸氢钠与碳酸铯, 但碳酸氢钠碱性较弱未反应, 碳酸铯碱性较强, 副产物较多, 并且a步反应中, 原药化合物B与溴代物C室温反应6 h即可, 但原药epacadostat与溴代物C需室温反应24 h。其中硝基呋喃类溴代物与原药反应时产率较低, 可能与硝基呋喃类前药化合物的稳定性有关。
| Table 1 Structures and physical property of the target compounds |
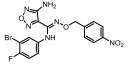
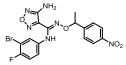
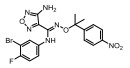
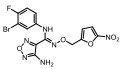
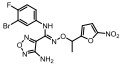
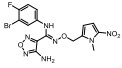
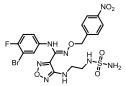
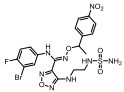
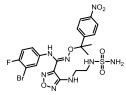
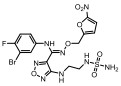
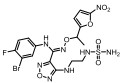
| Table 2 Spectral data of target compounds |
本文在化学合成、结构表征的基础上, 进一步对所合成的前药化合物进行了系统的评价。要求前药在体内到达肿瘤组织之前要相对稳定和低毒性, 在肿瘤组织缺氧微环境的作用下释放原药, 为此做了MTT实验以及目标化合物在PBS (0.01 mol·L-1, pH=7.4)缓冲溶液中与小鼠血浆中的稳定性实验。为了确定预期的前药在缺氧的肿瘤组织内是否能够释放出原药, 对合成的所有目标化合物进行了体外还原裂解筛选和评估, 并对个别毒性低、血浆中稳定、硝基还原酶环境下能够裂解的前药化合物进行了体内肿瘤组织中裂解的预判实验, 然后选择个别前药化合物与原药进行体内抗肿瘤活性比较。

|
Scheme 3 Process of degradation of the designed prodrug compound in tumor tissue to release the active drug |

|
Scheme 4 Synthetic route of target compounds. Reagents and conditions: a) K2CO3, DMF, rt, 6 h or 24 h |
体外细胞毒性实验结果(表 3)表明, 所合成前药化合物中F-1、F-4、F-5、F-6、F-10、F-11与原药细胞毒性相近, 但其中F-4、F-5、F-10、F-11可能与其稳定性较差有关(表 4), 其余前药化合物的细胞毒性略高于原药, 远低于阳性对照药, 符合前药化合物低毒性的要求。
| Table 3 The effect of IDO1 inhibitor prodrugs on the proliferation of cells. U87 MG: Human glioblastoma cell line; GL261: Mouse brain tumor cell line; HepG2: Human liver cancer cell line; H22: Mouse liver cancer cell line; LO2: Human liver cell strain. Positive control drug is 5-FU |
| Table 4 Stability test results in phosphate buffer solution (PBS) |
PBS稳定性实验结果(表 4)可以看出当A为苯环或吡咯环时, 如前药化合物F-1、F-2、F-3、F-6、F-7、F-8、F-9从0 h至31 h在37 ℃、PBS (0.01 mol·L-1, pH=7.4)缓冲溶液中都很稳定无裂解; 而当A为呋喃环时, 从第一次取样6 h时就已经检测不到前药化合物F-4、F-5、F-10、F-11, 所以为了确定前药化合物F-4、F-5、F-10、F-11的裂解时间, 再次将这4个化合物重新测定, 此次取样时间点为0.5、1、2和3 h。从结果(表 5)可以看出前药化合物F-4、F-5、F-10、F-11从0.5 h开始裂解, 随着时间的延长, 前药剩余的越来越少, 通过HPLC能够检测到有其他新的色谱峰产生。
| Table 5 Stability test results in PBS |
考虑到前药化合物在到达肿瘤之前要稳定, 进入肿瘤组织后再裂解释放原药较为理想。选取在PBS (0.01 mol·L-1, pH=7.4)缓冲溶液中稳定的F-1、F-2、F-3、F-6、F-7、F-8、F-9测定其体外酶还原实验情况。酶还原实验结果(表 6)表明:前药化合物能够释放出原药, 随着时间的延长, 前药含量越来越少, 原药含量逐渐增加, 其中前药化合物中A为对硝基苯的F-1在25 h内90%以上裂解为原药, 前药化合物中A为硝基吡咯环的F-6在25 h之内22%以上裂解为原药, 后续实验会继续观察。
| Table 6 Results of enzyme reduction experiment. The final concentration of nitroreductase is 5 µg·mL-1 |
综合PBS稳定性实验与体外硝基还原酶还原实验, 本实验从A为苯环、吡咯环中各选取1个在PBS中稳定性相对较好, 有代表性的, 并且在硝基还原酶还原实验中结果也较好的3个化合物F-1、F-6、F-7又进一步做了在小鼠血浆中的稳定性实验。
2.4 血浆中稳定性实验化合物在血浆中的稳定性实验结果(表 7)表明:在所设置的时间点内, 前药化合物F-1与F-7在血浆中较稳定, 与PBS中稳定性实验的结果也相对应。但在检测仪器中前药化合物F-7的响应较弱, 不明显。前药化合物F-6在血浆中3 h时之前相对稳定。所以选取化合物F-1与F-6测试前药化合物在小鼠肿瘤组织中能否被还原。
| Table 7 Results of plasma stability test. The control is 0.5% BSA in PBS (0.01 mol·L-1, pH=7.4) |
通过前药在小鼠肿瘤组织裂解的实验结果(表 8)发现:前药化合物F-1与F-6都能够在0.5 h内释放出大部分原药, 其中F-1在2 h后消失, F-6在1 h后消失, 虽然前药化合物F-6比F-1释药速度更快, 但是其在血浆中稳定性差。结果表明前药化合物F-1与F-6能够靶向到肿瘤组织中, 并且能够在肿瘤微环境低氧条件下通过酶的作用被还原, 释放出原药, 所以选择F-1与F-6进行体内药效学评价。
| Table 8 Results of reduction test of prodrug in mouse tumor tissue |
通过对小鼠H22肝癌的生长抑制作用结果(表 9)可以看出, 前药化合物F-1对肿瘤生长的抑制率高于原药组及阳性对照组5-FU, 而前药化合物F-6对肿瘤生长的抑制率与原药组相近, 低于阳性对照组。前药化合物F-1的抗肿瘤效果值得进一步研究。
| Table 9 The effect of prodrug compounds on the tumor growth of mouse H22 hepatocellular carcinoma xenograft model. *P < 0.05, **P < 0.01, ***P < 0.001 vs the solvent control group. Compound B is the original drug of the prodrug compounds F-1 and F-6 |
本研究基于肿瘤缺氧设计合成特异性靶向肿瘤组织的IDO1抑制剂前药, 探索其对肿瘤抑制的影响。基于报道的高活性IDO1抑制剂羟基脒类化合物B和epacadostat, 设计合成了11个前药化合物。通过MTT实验、稳定性实验、体外硝基还原酶还原实验、体内肿瘤组织缺氧降解实验, 从中选取出稳定性、释药速度等结果较好的化合物F-1与F-6用于药效学评价。体内初步药效评价结果表明, 前药F-1对肿瘤生长的抑制率明显高于原药组, 特异性抑制肿瘤组织的IDO1与全面抑制机体内IDO1的抗肿瘤效果明显不同, 进一步的药代分布和作用机制研究正在进行中。本研究将对今后的研究具有重要指导意义。
实验部分实验所用溶剂均为分析纯, 其中二氯甲烷经Innovative Technology PS-MD-7溶剂纯化系统干燥纯化后使用, 其他溶剂未特别指出则未经处理。所用试剂为化学纯或分析纯, 购买所得且未做进一步处理, 直接使用。核磁共振仪为Varian Mercury 400或500型, 内标为TMS。柱色谱分离用硅胶为青岛海洋化工厂生产的200~300目粗孔硅胶(zcx-Ⅱ), 薄层色谱硅胶为青岛海洋化工厂生产的硅胶GF254。
1 化学合成通法将化合物B/epacadostat (2 mmol)与碳酸钾(3 mmol)溶于DMF (10 mL), 室温搅拌30 min后, 将溴代物C (3 mmol)加入反应中, 搅拌过夜。加入水(20 mL)、乙酸乙酯(30 mL)萃取3次, 有机相用饱和食盐水洗涤, 无水硫酸钠干燥, 过滤, 减压浓缩后, 硅胶柱色谱分离得目标化合物。目标化合物理化性质和核磁数据见表 1、表 2。
2 生物学部分 2.1 IDO1前药对体外培养细胞的生长抑制作用实验将对数生长期的细胞用胰酶消化后, 配成每毫升2×104~3×104个的细胞悬液, 每孔100 μL加入96孔板中。次日加入不同浓度样品及相应溶剂对照的新鲜培养基, 每孔加100 µL, 受试化合物设4个剂量组, 每组设3个平行孔。于37 ℃培养箱继续孵育96 h后, 每孔加入新鲜配制的2 mg·mL-1 MTT溶液50 μL。继续培养4 h后弃上清液, 每孔加入DMSO 150 μL, 充分混匀后在波长570 nm处检测吸光度值, 以溶剂对照处理的肿瘤细胞为对照组, 按中效方程计算IC50。
2.2 PBS中稳定性实验将待测前药化合物与原药分别用DMSO配置成5 mmol·L-1的贮备液, 在5 mL的具塞刻度试管中加入DMSO 20 µL和贮备液4 µL, 用PBS (0.01 mol·L-1, pH=7.4)缓冲溶液稀释至2 mL, 涡旋30 s后于37 ℃恒温水浴锅孵育, 分别于6、24、31 h时取样500 µL, 按1:1比例加入甲醇500 µL, 涡旋2 min, 2 000 r·min-1离心5 min, 取上清液20 µL, 进行HPLC分析。
2.3 酶还原实验参考文献[16, 17]选择用硝基还原酶在体外模拟酶作用下的还原实验, 将待测前药化合物与原药分别用DMSO配置成5 mmol·L-1的贮备液, 将还原型烟酰胺腺嘌呤二核苷酸(NADH)用灭菌过的超纯水配置成50 mmol·L-1, 将硝基还原酶(NTR)用超纯水配置成0.5 µg·µL-1。在5 mL的具塞刻度试管中加入DMSO 25 µL、贮备液5 µL和PBS (0.01 mol·L-1, pH=7.4)缓冲溶液2.42 mL, 再加入配置好的NADH溶液25 µL与NTR溶液25 µL, 涡旋30 s后于37 ℃恒温水浴锅孵育, 分别于1、4、10、24 h时取样500 µL, 按1:1比例加入甲醇500 µL, 涡旋2 min, 2000 r·min-1离心5 min, 取上清液20 µL, 进行HPLC分析。
2.4 血浆中稳定性实验将待测前药化合物F-1、F-6、F-7用DMSO配置成5 mmol·L-1的贮备液, 再用含有0.5% BSA的PBS (0.01 mol·L-1, pH=7.4)磷酸盐缓冲溶液稀释为0.045 µg·µL-1。在5 mL的具塞刻度试管中加入小鼠血浆200 µL于37 ℃恒温振荡器中预热, 然后向其中加入浓度为0.045 µg·µL-1稀释后的贮备液100 µL。在另一5 mL的具塞刻度试管中加入0.5% BSA的PBS (0.01 mol·L-1, pH=7.4)磷酸盐缓冲溶液200 µL和浓度为0.045 µg·µL-1稀释后的贮备液100 µL作为对照组。另取一5 mL的具塞刻度试管中加入小鼠血浆200 µL与0.5% BSA的PBS (0.01 mol·L-1, pH=7.4)磷酸盐缓冲溶液100 µL作为空白对照。涡旋混匀5 min后于37 ℃恒温恒温振荡器中孵育, 分别于0.25、0.5、1、3、4.5 h时取样60 µL, 按1:2比例加入甲醇120 µL, 涡旋2 min, 2 000 r·min-1离心10 min, 取上清液20 µL, 进行HPLC分析。
2.5 前药在小鼠肿瘤组织的裂解购自中国食品药品检定研究院(批次号: 1112511800000798)的裸鼠, 体重16.0~18.0 g。无菌条件下, 剥离传代于裸鼠腋下的NCI-H1975和NCI-H460肺癌肿瘤组织, 去除筋膜, 剪碎瘤组织后分成大小均一的2~3 mm3瘤块, 接种于BALB/c裸鼠腋背部(左腋下接种NCI-H460, 右腋下接种NCI-H1975)。待肿瘤平均体积达到600 mm3时, 将动物随机分为2组, F-1给药组80 mg·kg-1和F-6给药组80 mg·kg-1, 腹腔注射, 给药后分0.5、1和2 h共3个时间点取材, 每个时间点1只。取材时, 处死动物, 剥离肿瘤组织, 加入生理盐水匀浆(肿瘤组织:生理盐水=1:3), 涡旋3 min后2 000 r·min-1离心5 min后取上清液300 µL, 加入300 µL乙腈, 涡旋1 min, 2 000 r·min-1离心5 min, 重复两次后, 取上清液20 µL进行HPLC分析。所有的动物实验均严格遵循中国医学科学院药物研究所动物实验中心标准操作规程, 减少动物实验对动物造成的伤害。
2.6 前药化合物对小鼠H22肝癌的生长抑制作用小鼠肝癌H22模型, 选用的动物为KM小鼠, 体重15.0~18.0 g (由北京市中国药品生物制品检定所实验动物中心提供)。取生长良好的腹水, 用无菌生理盐水按1:3比例稀释后制成肿瘤细胞悬液, 每只小鼠腋背部接种0.2 mL瘤液。接种后次日动物随机分组, 称重。实验动物共分5组, 荷瘤溶剂对照组、阳性对照药5-FU给药组30.0 mg·kg-1、F-1给药组100 mg·kg-1、原药给药组100 mg·kg-1、F-6给药组100 mg·kg-1, 每组5只动物。5-FU给药组30.0 mg·kg-1于接种后第1、4、7天分别给药1次, 共给药3次, 给药体积为每20 g小鼠腹腔注射0.2 mL, 溶剂对照、原药与受试化合物F-1和F-6给药体积为每20 g小鼠灌胃0.4 mL, 每日给药2次, 于接种后第4天开始给药, 共给药5天。
实验结束后, 颈椎脱臼处死动物, 称体重, 剥取肿瘤组织并称重。根据重量计算肿瘤抑制率(%)、体重、瘤重用均值±标准差(± SD)表示, 并进行各给药组与溶剂对照组之间的t检验。所有的动物实验均严格遵循中国医学科学院药物研究所动物实验中心标准操作规程, 减少动物实验对动物造成的伤害。
| [1] |
Macchiarulo A, Camaioni E, Nuti R. Highlights at the gate of tryptophan catabolism:a review on the mechanisms of activation and regulation of indoleamine 2, 3-dioxygenase (IDO), a novel target in cancer disease[J]. Amino Acids, 2009, 37: 219-229. DOI:10.1007/s00726-008-0137-3 |
| [2] |
Pang WQ, Gao L, Dou YY, et al. Screening of natural small molecule IDO-1 inhibitors with anti-tumor role[J]. Acta Pharm Sin (药学学报), 2017, 52: 1416-1423. |
| [3] |
Platten M, von Knebel Doeberitz N, Oezen I, et al. Cancer immunotherapy by targeting IDO1/TDO and their downstream effectors[J]. Front Immunol, 2015, 5: 673-679. |
| [4] |
Liu X, Shin N, Koblish HK, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity[J]. Blood, 2010, 115: 3520-3530. DOI:10.1182/blood-2009-09-246124 |
| [5] |
Moon YW, Hajjar J, Hwu P. Targeting the indoleamine 2, 3-dioxygenase pathway in cancer[J]. J Immunother Cancer, 2015, 3: 51-54. DOI:10.1186/s40425-015-0094-9 |
| [6] |
Du TT, Lai FF, Chen XG. Research progress of indoleamine 2, 3-dioxygenase 1 in tumor immunotherapy[J]. Acta Pharm Sin (药学学报), 2018, 53: 1271-1278. |
| [7] |
Long GV, Dummer R, Hamid O. Epacadostat (E) plus pembrolizumab (P) versus pembrolizumab alone in patients (pts) with unresectable or metastatic melanoma:results of the phase 3 ECHO-301/KEYNOTE-252 study[J]. J Clin Oncol, 2018(15_suppl): 108-108. DOI:10.1200/JCO.2018.36.15_suppl.108 |
| [8] |
Wainwright DA, Balyasnikova IV, Chang AL. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival[J]. Clin Cancer Res, 2012, 18: 6110-6121. DOI:10.1158/1078-0432.CCR-12-2130 |
| [9] |
Wainwright DA, Chang AL, Dey M. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors[J]. Clin Cancer Res, 2014, 20: 5290-5301. DOI:10.1158/1078-0432.CCR-14-0514 |
| [10] |
Wei X, Chen X, Ying M, et al. Brain tumor-targeted drug delivery strategies[J]. Acta Pharm Sin B, 2014, 4: 193-201. DOI:10.1016/j.apsb.2014.03.001 |
| [11] |
Wilson WR, Hay MP. Targeting hypoxia in cancer therapy[J]. Nat Rev Cancer, 2011, 11: 393. DOI:10.1038/nrc3064 |
| [12] |
Ikeda Y, Hisano H, Nishikawa Y. Targeting and treatment of tumor hypoxia by newly designed prodrug possessing high permeability in solid tumors[J]. Mol Pharm, 2016, 13: 2283-2289. DOI:10.1021/acs.molpharmaceut.6b00011 |
| [13] |
Boer J, Young-Sciame R, Lee F. Roles of UGT, P450, and gut microbiota in the metabolism of epacadostat in humans[J]. Drug Metab Dispos, 2016, 44: 1668-1674. DOI:10.1124/dmd.116.070680 |
| [14] |
Tao M, Frietze W, Meloni DJ, et al. Process for the synthesis of an indoleamine 2, 3-dioxygenase inhibitor: US 14/535, 781[P]. 2015-5-14.
|
| [15] |
Winn BA, Shi Z, Carlson GJ. Bioreductively activatable prodrug conjugates of phenstatin designed to target tumor hypoxia[J]. Bioorg Med Chem Lett, 2017, 27: 636-641. DOI:10.1016/j.bmcl.2016.11.093 |
| [16] |
O'Connor LJ, Cazares-Körner C, Saha J. Design, synthesis and evaluation of molecularly targeted hypoxia-activated prodrugs[J]. Nat Protoc, 2016, 11: 781-794. DOI:10.1038/nprot.2016.034 |
| [17] |
Li Y, Sun Y, Li J. Ultrasensitive near-infrared fluorescence-enhanced probe for in vivo nitroreductase imaging[J]. J Am Chem Soc, 2015, 137: 6407-6416. DOI:10.1021/jacs.5b04097 |