 2019, Vol. 54
2019, Vol. 54



由于临床中抗生素的长期使用, 导致细菌发生变异和耐药, 且其在医疗和农业生产中的不当使用加剧了细菌耐药的发生, 并已逐渐成为威胁人类健康的重大问题。2017年世界卫生组织公布了最需要抗生素的12种细菌或细菌家族清单[1] (表 1), 其中革兰氏阴性菌中的鲍曼不动杆菌(Acinetobacter baumannii)、铜绿假单胞菌(Pseudomonas aeruginosa)和产广谱β-内酰胺酶(extended spectrum beta-lactamases, ESBLs)的肠杆菌(Enterobacteriaceae)处于第一优先级, 此外还有6种革兰氏阴性菌也列于名单之上, 如此严峻的形势使得抗革兰氏阴性菌药物的研究刻不容缓。革兰氏阴性菌最显著的特征是它们的细胞膜由内膜、含有较薄肽聚糖层的水性周质和外膜组成, 正是由于其结构上的特殊性, 使得大部分抗生素很难穿过细胞膜产生抗菌作用。此外革兰氏阴性菌还具有外排泵, 药物进入后会被感知并排出细胞外, 因此, 抗革兰氏阴性菌的药物研发非常困难。本文通过对现有研究进行总结, 按照不同作用机制对目前抗革兰氏阴性菌的化合物研究情况进行简要的综述, 为设计、合成新型抗革兰氏阴性菌药物提供一定参考。
| Table 1 WHO priority pathogens list for R & D of new antibiotics[1] |
革兰氏阴性菌的细胞膜由内膜、含有较薄肽聚糖层的水性周质和外膜组成。外膜具有独特的组成, 包括脂蛋白、β-barrel蛋白、脂多糖和磷脂。其中脂蛋白是一种膜蛋白, 具有多种功能, 如细胞表面的合成和维护以及底物的运输等。脂蛋白在细胞质中合成, 然后由脂蛋白转运系统(lipoprotein transport system, Lol system)转运到外膜。Lol系统包含5种蛋白, LolCDE作为一个ATP结合盒(ATP binding cassette, ABC)转运体复合物, 可以使脂蛋白从内膜上脱离后与LolA在周质形成水溶性复合物, 脂蛋白随后被转移到外膜上的LolB, 进而参与形成磷脂层(图 1)[2, 3], 阻断此途径, 可以使细胞变形、溶解进而达到杀菌目的。

|
Figure 1 Transport of lipoproteins by the Lol system in E. coli. |
2015年McLeod等[3]通过高通量表型筛选, 首次发现通过作用于LolCDE转运体复合物来阻断蛋白转运, 从而发挥抗菌作用的化合物1和2 (图 2), 化合物的结构骨架为吡啶咪唑。化合物2对大肠杆菌和流感嗜血杆菌的抗菌活性高于1, MIC降低4~8倍。Nayar等[4]通过高通量筛选, 也发现一个结构类型为吡唑的LolCDE转运体抑制剂3, 该化合物对于外排突变型和野生型大肠杆菌的MIC分别为0.125 μg·mL-1 (Levofloxacin: 0.004 μg·mL-1)和8 μg·mL-1。

|
Figure 2 Inhibitors of LolCDE transporter |
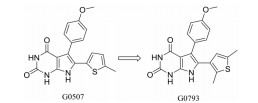
Nickerson等[5]通过表型筛选, 发现一个骨架结构为吡咯嘧啶二酮的新型LolCDE转运体抑制剂G0507, 并对其进行了结构改造(表 2)。其中化合物G0793对大肠杆菌imp4213和大肠杆菌ΔtolC的MIC分别为0.25 μg·mL-1和0.125 μg·mL-1。
| Table 2 Antibacterial activities of compounds G0507 and G0793 |
革兰氏阴性菌的外膜是含有脂多糖(lipopolysaccharide, LPS)的不对称磷脂双分子层, 为革兰氏阴性菌特有结构, 是维持其形态以及保证细胞通透性的屏障。因此可以通过抑制LPS的形成来破坏屏障, 使细胞无法维持正常形态, 以达到杀死革兰氏阴性菌的目的。
1.2.1 阻断脂多糖转运系统脂多糖转运系统(lipopolysaccharide transport system, Lpt system)负责将LPS从内膜转移至外膜, 由7个蛋白组成: LptA、B、C、D、E、F和G, 这7个蛋白被划分成3个区域:内膜复合物LptBFGC、周质蛋白LptA以及外膜复合物LptD/E (图 3)[6, 7]。Luo等[8]报道了来源于铜绿假单胞菌的LptB2FG晶体结构, 功能分析表明通过LptB与ATP结合和水解诱导的构象改变, LPS可能横向进入由LptF和LptG的跨膜区组成的Ⅴ型中心腔, 然后进一步排入LptF和LptG周质β-卷曲区域。这些研究表明, LptB2FG提取LPS的机制不同于经典的ABC转运蛋白。

|
Figure 3 Export of LPS to the cell surface |
氨基香豆素类化合物新生霉素(novobiocin)是一个疏水的DNA旋转酶抑制剂(图 4), 由于安全因素已被撤回。但研究发现其可以提高LptB2FG的活性, 加快后续的LPS转运[9], 在以LPS从内膜上排出作为靶点时, 可以考虑将新生霉素作为潜在的先导化合物进行合理设计进而产生抑制作用[10]。

|
Figure 4 Novobiocin and IMB-881 |
LptC对于LPS的转运和细胞存活至关重要, LptC与LptB2FG结合于内膜外侧, 为LptA提供结合位点, 并将LPS传递给LptA[11-13]。LptA携带LPS穿过细胞周质递送至外膜上的LptD/E, 通过LptD/E传送至膜外, 形成LPS层, 完成LPS的转运[14-16]。
Zhang等[17]以大肠杆菌中LptC和LptA的相互作用为基础建立一种筛选系统, 用该系统验证化合物IMB-881 (图 4)通过干扰LptC和LptA相互作用来抑制革兰氏阴性菌的生长, 对临床耐碳青霉烯的大肠杆菌和临床多药耐药的鲍曼不动杆菌的MIC分别为6.25~25 μg·mL-1和12.5~50 μg·mL-1。
截至目前, LptD/E复合物将LPS传送至外膜的机制还不清楚, 但由于LptD/E复合物位于外膜处, 更易接近, 所以相对于其他蛋白是更加有前景的靶点[10]。大环肽模拟物murepavadin (POL7080)和L27-11 (图 5)在铜绿假单胞菌中可直接与LptD作用来抑制LPS的形成, 二者对铜绿假单胞菌ATCC 27853的MIC分别为0.008和0.01 μg·mL-1, 对铜绿假单胞菌PAO1菌株的MIC均为0.004 μg·mL-1 (Ciprofloxacin: 0.5 μg·mL-1)[18, 19], 由Polyphor公司开发的POL7080已经进入Ⅲ期临床研究。

|
Figure 5 Murepavadin and L27-11 |
LPS由3部分组成, 延伸的氧抗原链、核心低聚糖区域以及磷酸化二糖脂质, 即类脂A。LPS通过类脂A锚定在细胞膜上, 因此类脂A的生物合成在革兰氏阴性菌生长过程中起着极其重要的作用。Raetz等首先提出类脂A的生物合成途径[20-22], 即Raetz pathway (合成路线1)。在此通路中, 由于UDP-3-O-(R-羟基十四酰)-N-乙酰氨基葡萄糖脱乙酰酶(LpxC)与任何哺乳动物蛋白都没有同源性且其催化的去乙酰化反应是合成类脂A的关键步骤, 为非常有前景的靶点。

|
Scheme 1 Raetz pathway |
大部分LpxC抑制剂包含可与Zn离子结合的头部基团以及可占据疏水口袋的亲脂性尾部基团(图 6)。亲脂性尾部与LpxC之间通过范德华力相互作用, 是抑制剂发挥活性的主要原因。Chiron公司发现的化合物CHIR-090是目前研究最充分的LpxC抑制剂, 对大部分革兰氏阴性菌均具有显著抗菌活性, 其中对大肠杆菌和铜绿假单胞菌的MIC90分别为0.25 μg·mL-1和4 μg·mL-1 (Meropenem: > 64 μg·mL-1)[23, 24]。LPC-058和CHIR-090同属于苏氨酰异羟肟酸衍生物, LPC-058是目前为止体外抗菌活性最强的化合物, 并且对产VIM-1碳青霉烯酶的铜绿假单胞菌和产OXA-23碳青霉烯酶的鲍曼不动杆菌依然有效, MIC均为0.5 μg·mL-1 (Ciprofloxacin: 16 μg·mL-1; 32 μg·mL-1)[25, 26]。化合物PF-5081090 (LpxC-4)结构骨架为吡啶酮甲基砜异羟肟酸[27], 也展现出较强的体外抗菌活性, 对大肠杆菌和铜绿假单胞菌的MIC90分别为0.25 μg·mL-1和1 μg·mL-1 (Meropenem: > 64 μg·mL-1)[23]。上述化合物目前均处于临床前研究阶段。Achaogen公司开发的丁二炔衍生物ACHN-975对大肠杆菌ATCC25922菌株、铜绿假单胞菌ATCC27853菌株的MIC分别为0.125 μg·mL-1和0.25 μg·mL-1[28], 是唯一一个进入临床研究的LpxC抑制剂, 虽然由于注射部位的炎症反应而停止了Ⅰ期试验, 但其仍然是LpxC抑制剂研究的一个突破, 为后续抗革兰氏阴性菌药物的研发提供了指导。

|
Figure 6 Inhibitors of LpxC |
组成LPS的核心低聚糖区可进一步分为:由3-脱氧-D-甘露-2-辛酮糖酸(3-deoxy-D-manno-oct-2-ulosonic acid, Kdo)和庚糖残基组成的内部核心, 由己糖衍生物组成的外部核心。细胞存活所必需的LPS结构为ReLPS, ReLPS仅由类脂A和Kdo残基组成。通过抑制庚糖的合成或转移可以阻止庚糖与类脂A以及Kdo2形成复合物(合成路线2), 从而形成ReLPS以达到抗菌目的, 因此抑制庚糖的生物合成或转移是非常有研究价值的抗菌靶点[29]。
Moreau等[30]通过虚拟筛选发现了首个到达微摩尔级别的庚糖转移酶WaaC的抑制剂, 通过结构优化, 发现化合物4 (图 7)对WaaC的IC50为1 μmol·L-1。随后, 该研究团队又对庚糖生物合成途径中的双功能酶HldE进行了研究。大肠杆菌的HldE酶由HldE激酶和腺苷转移酶两部分组成, 通过高通量筛选和构效关系研究发现了化合物5, 它对HldE激酶的IC50值为0.11 μmol·L-1[29]。然而化合物5对HldE激酶的抑制活性虽然很高, 但对细菌的抗菌能力却并不能令人满意, 于是该团队又进行了二次筛选, 发现了化合物6, 其对HldE激酶的IC50值为6 μmol·L-1。对化合物6进行构效关系研究后, 首次鉴定到对选定大肠杆菌菌株有抑制作用的化合物, 其中化合物7对大肠杆菌HldE激酶的IC50值为0.049 μmol·L-1, 与1 μg·mL-1红霉素联用时对大肠杆菌K12 Δ-acrAB, Δ-tolC的MIC值为2 μg·mL-1, 对大肠杆菌K1 Δ-tolC的MIC值为16 μg·mL-1[31]。

|
Scheme 2 Synthesis of ADP-L-glycero-β-D-manno-heptose from sedoheptulose-7-phosphate and incorporation into LPS |

|
Figure 7 Inhibitors of WaaC and HldE |
拓扑异构酶是存在于细胞核内的一类酶, 能够催化DNA链的断裂和结合, 从而控制DNA的拓扑状态, 在生物体内发挥重要作用。大部分原核生物的Ⅱ型拓扑异构酶均可进一步分为两个亚类: DNA旋转酶和Ⅳ型拓扑异构酶。DNA旋转酶为A、B两个亚基组成的GyrA2GyrB2四聚体, Ⅳ型拓扑异构酶为C、E两个亚基组成的ParC2ParE2四聚体[32]。GyrA和ParC两个亚基内含DNA链断裂位点, GyrB和ParE两个亚基内含ATP酶域, 通过水解ATP来为酶发挥作用提供必需的能量[33]。
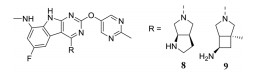
由于两个酶结构的高度相似性, Ⅱ型拓扑异构酶抑制剂通常为DNA旋转酶和Ⅳ型拓扑异构酶双靶点抑制剂, 可以通过阻断两者的ATP结合位点或催化过程来发挥抗菌作用。Tari等[34]发现了以嘧啶并吲哚为核心的三环类化合物, 可与DNA旋转酶和Ⅳ型拓扑异构酶的ATP结合位点结合发挥抗菌作用。其中化合物8和9对临床常见的革兰氏阴性菌具有强效的抗菌活性(表 3)。
| Table 3 Compounds and antibacterial activity |
氟喹诺酮类作为在DNA呈断裂状态时与Ⅱ型拓扑异构酶结合的药物, 在过去几十年内一直是该领域的代表[35]。杨森制药发现一个新的氟喹诺酮类化合物JNJ-Q2 (图 8), 目前由Furiex制药接手进行后续研究。该化合物抗革兰氏阴性菌的活性与莫西沙星相当, 但具有更好的药代动力学性质[36], 对铜绿假单胞菌、耐环丙沙星的大肠杆菌、流感嗜血杆菌和肺炎克雷伯菌的MIC分别为0.5~4、1~16、0.008~0.015和0.015~1 μg·mL-1 (Moxifloxacin: 0.5~8、8~ > 256、0.008~0.06和0.015~1 μg·mL-1)[37]。

|
Figure 8 Inhibitors of type Ⅱ topoisomerase |
虽然对氟喹诺酮类化合物的母核进行改造修饰的研究很多, 却没有人将3位羧基去掉, 这是由于3位羧酸和4位羰基被认为是发挥活性的药效团。Chu等[38, 39]用异噻唑-3(2H)-酮替代3位羧基, 设计合成了2, 3, 4, 9-四氢异噻唑[5, 4-b]喹啉-3, 4-二酮类化合物。该类化合物展现出强效抗菌活性, 为喹诺酮类药物的改造提供了更大的空间。随后, 一个异噻唑喹啉酮类化合物ACH-702 (图 8)被Achillion制药公司开发[36]。ACH-702对流感嗜血杆菌展现出超强活性, MIC为0.06~0.12 μg·mL-1 (Ampicillin: 0.12~ > 16 μg·mL-1)[40]。
近年来, 研究人员报道了一些结构类型和作用机制不同于氟喹诺酮类的新型拓扑异构酶抑制剂(novel bacteria topoisomerase inhibitors, NBTIs)。不同于氟喹诺酮类药物在DNA呈断裂状态时与酶结合, NBTIs在DNA呈完整状态时与酶结合[41]。2010年葛兰素史克的研究人员通过表型筛选发现一个哌啶烷喹啉类Ⅱ型拓扑异构酶抑制剂GSK-299423 (图 8), 通过对DNA旋转酶-底物(DNA)-GSK-299423复合物的晶体结构进行研究, GSK-299423与DNA旋转酶的结合位点不同于喹诺酮类化合物, 在X-衍射晶体结构中, DNA旋转酶处于还未开始切割DNA的阶段, 这是第一次捕捉到DNA旋转酶的这种构象[41]。该化合物对革兰氏阳性菌和阴性菌均有很强的活性, 对大肠杆菌、流感嗜血杆菌、肺炎克雷伯菌和铜绿假单胞菌的MIC值分别为0.063、0.25、0.5和8 μg·mL-1 (Ciprofloxacin: 0.004、≤0.004、0.016和0.5 μg·mL-1), 并且能够显著降低hERG毒性(IC50 = 1.4 mmol·L-1)[36, 41]。
Dougherty等[42]也发现一个新型拓扑异构酶抑制剂NBTI 5463 (图 8), 具有强效的抗革兰氏阴性菌活性。对部分革兰氏阴性菌的MIC50值为:大肠杆菌= 1 μg·mL-1, 铜绿假单胞菌= 0.5 μg·mL-1, 鲍曼不动杆菌= 8 μg·mL-1, 肺炎克雷伯菌= 4 μg·mL-1 (Cefepime: ≤0.06、8、16和0.25 μg·mL-1)。
另一类此作用机制的化合物结构骨架为苯并异噁唑, 阿斯利康公司通过对其进行构效关系研究发现, 噁唑烷酮取代的化合物避免了骨髓抑制毒性和基因毒性, 从而发现了化合物ETX0914[43] (图 8)。该化合物对耐喹诺酮的奈氏淋球菌(N. gonorrhoeae) ARC4676菌株有较强的活性, MIC值为0.125 μg·mL-1 (Ciprofloxacin: 32 μg·mL-1)[44], 已完成Ⅱ期临床试验即将开展Ⅲ期临床试验。
1.4 丝状温敏蛋白Z抑制剂丝状温敏蛋白Z (filamentous temperature-sensitive protein Z, FtsZ)是一个依赖于GTP的原核生物细胞分裂蛋白, 与真核生物细胞分裂蛋白微管蛋白的序列一致性低于10%, 但二者具有高度的结构相似性[45]。由于革兰氏阴性菌结构的特殊性, 目前已知的FtsZ抑制剂大多数具有抗革兰氏阳性菌活性, 通过文献调研, 发现几个对革兰氏阴性菌有抑制活性的化合物。
从血红根(Sanguinaria canadensis)的根茎中提取出的苯菲啶类天然产物血根碱(sanguinarine) (图 9)对大部分的革兰氏阳性菌和阴性菌均有抗菌活性, 对大肠杆菌BL21 (野生型)和JM109 (WM647)的IC50分别为4.6 ± 0.8和12.0 ± 1.7 μg·mL-1[46]。

|
Figure 9 Inhibitors of FtsZ |
Hogan等[47]利用测序技术发现一个新型苯并噻二唑衍生物C109 (图 9)。经后续形态筛选、验证以及荧光显微观察确定C109为FtsZ抑制剂。具有抗革兰氏阴性菌活性, 对鲍曼不动杆菌 ATCC 19606、大肠杆菌117782、大肠杆菌120955的MIC值分别为16、4、8 μg·mL-1。
Sun等[48]基于细胞筛选方法发现Ftsz抑制剂化合物10 (图 9), 对大肠杆菌ATCC 25922、大肠杆菌ATCC BAA-2469、铜绿假单胞菌ATCC BAA-2108、肺炎克雷伯菌ATCC BAA-1144、鲍曼不动杆菌ATCC 19606的MIC值分别为4、4、16、16、16 μg·mL-1 (Methicillin: 2、> 1 024、> 256、> 256、> 256 μg·mL-1)。同时该化合物对哺乳动物细胞的细胞毒性很低, 是一个具有前景的先导化合物。
1.5 β内酰胺酶(β-lactamase)抑制剂β内酰胺类抗生素是第一个被成功开发的天然抗菌剂, 由于其良好的抗菌活性和选择性, 至今美国临床可注射抗生素超过65%仍为该类抗生素[49]。β内酰胺酶的产生是该类抗生素产生耐药的主要原因, 为了克服这种耐药性, 除了研制具有耐酶性能的新抗生素外, 还要不断寻找新的β内酰胺酶抑制剂, 通过将β内酰胺酶抑制剂与β内酰胺类抗生素联用来发挥抗菌作用。
LN-1-255 (图 10)是由Pattanaik等[50]设计的β内酰胺类β内酰胺酶抑制剂, 具有强效抗菌活性。它可以显著提高头孢他啶(ceftazidime)和头孢匹罗(cefpirome)对临床耐碳青霉烯菌株的抗菌活性。与碳青霉烯类抗生素联用可以有效抑制鲍曼不动杆菌D型碳青霉烯酶, 在根除携带CHDLs的鲍曼不动杆菌引起的感染中起到重要作用[51]。

|
Figure 10 Inhibitors of β-lactamase |
首个非β内酰胺类的β内酰胺酶抑制剂阿维巴坦(avibactam)于2015年获得美国FDA批准, 与头孢他啶按4:1的比例联合用药, 可治疗复杂尿路感染。阿维巴坦是二氮杂双环辛酮(diazabicyclooctonane, DBOs)类化合物, 继它之后又陆续推出几个该类型化合物, 均到达临床试验阶段, 包括relebactam、zidebactam、ETX2514以及nacubactam (图 10)[52]。
与阿维巴坦结构不同的vaborbactam (图 10)是非β-内酰胺结构的环硼酸, 硼原子作为亲电试剂可以与特定β-内酰胺酶的催化位点丝氨酸形成可逆的共价结合[53]。与美罗培南(meropenem)联用组成的Vabomere由The Medicines公司开发, 已于2017年8月底获美国FDA批准, 该药作用于产β-内酰胺酶的革兰氏阴性菌, 用于成人患者复杂性尿路感染和肾盂肾炎的治疗[54, 55]。
VenatoRx制药公司研发的VNRX-5133 (图 10)与vaborbactam结构相似, 它对丝氨酸-β-内酰胺酶和头孢菌素酶都具有活性, 可与头孢吡肟(cefepime)联用治疗革兰氏阴性菌感染, 目前已进入临床Ⅰ期试验[56, 57]。
1.6 信号肽酶Ⅰ抑制剂细菌中超过30%的蛋白需要在细胞膜或者细胞外膜发挥不同功能, 如摄取营养, 产生毒性或起到防御作用。这些蛋白在发挥作用过程中需要通过细菌中普遍存在的分泌途径来进行运输。在分泌途径中, 信号肽和蛋白结合形成前蛋白, 然后将前蛋白转移至质膜上, 通过信号肽酶加工使信号肽从蛋白上脱离, 将蛋白释放到膜外发挥作用。大部分的前蛋白都是经由信号肽酶Ⅰ (type Ⅰ signal peptidase, SPase Ⅰ)加工, 若抑制此酶可使前蛋白在质膜上累积, 导致细胞死亡, 因此, SPase Ⅰ为很有研究价值的抗菌药物靶点[58]。
Arylomycin是大环脂肽类SPase Ⅰ抑制剂, Smith等[59]为了增强arylomycin类抑制剂对靶标的亲和力以及透膜能力, 以arylomycin A-C16为先导化合物进行结构优化, 发现了化合物G0775 (图 11), 该化合物具有强效、广谱抗革兰氏阴性菌能力, 对MDR菌株也有很强的体外活性, MIC值在0.125~2 μg·mL-1 (arylomycin A-C16: > 64 μg·mL-1), 并且对大肠杆菌、肺炎克雷伯菌、铜绿假单胞菌和鲍曼不动杆菌具有较强的体内杀菌活性。相关的研究结果表明化合物G0775是一个非常具有前景的抗革兰氏阴性菌候选药物。

|
Figure 11 Inhibitors of SPase Ⅰ |
铁的获取是细菌建立感染周期的重要步骤[60]。微生物有多种方式来获取铁, 其中一种是细菌通过将小分子量、高亲和力的铁载体分泌到细胞外, 在外界摄取铁后再通过位于细胞外膜的铁载体转运到细胞内将铁释放[61]。外膜通透性降低是革兰氏阴性菌产生耐药性的主要原因之一, 可以采用特洛伊木马方法来解决此问题。特洛伊木马就是通过linker将铁载体和抗生素结合, 利用革兰氏阴性菌对铁的摄取从而将铁载体-抗生素复合物转运至细胞内, 使得抗生素可以进入细胞内发挥作用[61, 62]。
β内酰胺类药物的靶标青霉素结合蛋白(penicillin binding proteins, PBPs)位于细胞周质, 细胞外膜是该类药物进入细胞的唯一屏障, 因此该类抗菌药非常适合特洛伊木马策略[62], cefiderocol (图 12)就是一个成功的例子, 该化合物是与邻苯二酚型铁载体模拟物相结合的头孢菌素类抗生素, 显著提高了对于细菌细胞的渗透能力以及活性, 使得它对于碳青霉烯抗性杆菌如鲍曼不动杆菌和铜绿假单胞菌也有很好的抗菌作用[63], 已完成临床试验并提交上市申请。

|
Figure 12 Structures of siderophore-conjugated β-lactam antibiotics |
在所有的β内酰胺类抗生素中, 单环类β内酰胺抗生素由于强效的抗革兰氏阴性菌活性以及对头孢菌素酶在内的β内酰胺酶具有抗性的特点而更适合做成铁载体模拟物结合型抗生素[64]。
BAL30072 (图 12)是Basilea制药公司发现的monosulfactam类铁载体模拟物结合型β内酰胺抗生素。该化合物对多药耐药的铜绿假单胞菌和鲍曼不动杆菌(包括耐碳青霉烯菌株)具有强效抑制作用, 已进入临床Ⅰ期试验阶段[65, 66]。Tan等[67]通过对BAL30072的铁载体模拟物以及linker等部分进行修饰, 合成了一系列活性较好的化合物, 其中化合物11 (图 12)体外抗革兰氏阴性菌活性强, 血浆蛋白结合率适当并且稳定性良好, 具有优良的代谢性质和较强的体内抗菌活性(表 4)。
| Table 4 Compound 11 and biological activities. aEco, E. coli, 5 strains; Kpn, MDR K. pneumoniae, 5 strains; Aba, MDR A. baumannii, 5 strains; Pae, MDR P. aeruginosa, 5 strains. bn = 4, data represent as mean ± standard deviation; fu, Fraction unbound. cStability: The degradation percentage after 4 h incubation in human plasma at 37 ℃. d Sprague-Dawley rat (male), 1 mg·kg-1, n = 3 |
由于多药耐药菌和广泛耐药菌的大量出现, 新型抗生素的研发迫在眉睫。革兰氏阴性菌具有特殊的生物学结构, 使得抗革兰氏阴性菌药物一直以来都是抗菌药物研发中的难点。本综述以不同的作用机制为切入点介绍了具有良好抗革兰氏阴性菌活性的化合物。虽然大部分化合物处于生物活性研究或临床前研究阶段, 进入临床试验阶段的化合物还比较少, 未来还具有很多的不确定性, 但这些具有不同作用机制的不同结构类型化合物的出现为药物研发人员提供了宝贵的经验和新的研究思路, 使得新型抗菌药物的出现成为可能。此外, 近年来结构生物学特别是冷冻电镜技术的发展, 相信将有更多的与革兰氏阴性菌有关的靶蛋白结构被解析出来, 这将进一步加速新型抗革兰氏阴性菌药物的研发。
| [1] | World Health Organization. WHO publishes list of bacteria for which new antibiotics are urgently needed [N]. Geneva: WHO, 2017. |
| [2] | Narita S. ABC transporters involved in the biogenesis of the outer membrane in gram-negative bacteria[J]. Biosci Biotechnol Biochem, 2011, 75: 1044–1054. DOI:10.1271/bbb.110115 |
| [3] | McLeod SM, Fleming PR, MacCormack K, et al. Small-molecule inhibitors of gram-negative lipoprotein trafficking discovered by phenotypic screening[J]. J Bacteriol, 2015, 197: 1075–1082. DOI:10.1128/JB.02352-14 |
| [4] | Nayar AS, Dougherty TJ, Ferguson KE, et al. Novel antibacterial targets and compounds revealed by a high-throughput cell wall reporter assay[J]. J Bacteriol, 2015, 197: 1726–1734. DOI:10.1128/JB.02552-14 |
| [5] | Nickerson NN, Jao CC, Xu Y, et al. A novel inhibitor of the LolCDE ABC transporter essential for lipoprotein trafficking in gram-negative bacteria[J]. Antimicrob Agents Chemother, 2018, 62: e02151–17. |
| [6] | Sperandeo P, Lau FK, Carpentieri A, et al. Functional analysis of the protein machinery required for transport of lipopolysaccharide to the outer membrane of Escherichia coli[J]. J Bacteriol, 2008, 190: 4460–4469. DOI:10.1128/JB.00270-08 |
| [7] | Laguri C, Sperandeo P, Pounot K, et al. Interaction of lipopolysaccharides at intermolecular sites of the periplasmic Lpt transport assembly[J]. Sci Rep, 2017, 7: 9715. DOI:10.1038/s41598-017-10136-0 |
| [8] | Luo Q, Yang X, Yu S, et al. Structural basis for lipopolysaccharide extraction by ABC transporter LptB2FG[J]. Nat Struct Mol Biol, 2017, 24: 469–474. DOI:10.1038/nsmb.3399 |
| [9] | May JM, Owens TW, Mandler MD, et al. The antibiotic novobiocin binds and activates the ATPase that powers lipopolysaccharide transport[J]. J Am Chem Soc, 2017, 139: 17221–17224. DOI:10.1021/jacs.7b07736 |
| [10] | Hicks G, Jia Z. Structural basis for the lipopolysaccharide export activity of the bacterial lipopolysaccharide transport system[J]. Int J Mol Sci, 2018, 19: E2680. DOI:10.3390/ijms19092680 |
| [11] | Narita S, Tokuda H. Biochemical characterization of an ABC transporter LptBFGC complex required for the outer membrane sorting of lipopolysaccharides[J]. FEBS Lett, 2009, 583: 2160–2164. DOI:10.1016/j.febslet.2009.05.051 |
| [12] | Sperandeo P, Villa R, Martorana AM, et al. New insights into the Lpt machinery for lipopolysaccharide transport to the cell surface: LptA-LptC interaction and LptA stability as sensors of a properly assembled transenvelope complex[J]. J Bacteriol, 2011, 193: 1042–1053. DOI:10.1128/JB.01037-10 |
| [13] | Suits MD, Sperandeo P, Deho G, et al. Novel structure of the conserved gram-negative lipopolysaccharide transport protein A and mutagenesis analysis[J]. J Mol Biol, 2008, 380: 476–488. DOI:10.1016/j.jmb.2008.04.045 |
| [14] | Qiao S, Luo Q, Zhao Y, et al. Structural basis for lipopolysaccharide insertion in the bacterial outer membrane[J]. Nature, 2014, 511: 108–111. DOI:10.1038/nature13484 |
| [15] | Chng SS, Ruiz N, Chimalakonda G, et al. Characterization of the two-protein complex in Escherichia coli responsible for lipopolysaccharide assembly at the outer membrane[J]. Proc Natl Acad Sci U S A, 2010, 107: 5363–5368. DOI:10.1073/pnas.0912872107 |
| [16] | Freinkman E, Chng SS, Kahne D. The complex that inserts lipopolysaccharide into the bacterial outer membrane forms a two-protein plug-and-barrel[J]. Proc Natl Acad Sci U S A, 2011, 108: 2486–2491. DOI:10.1073/pnas.1015617108 |
| [17] | Zhang X, Li Y, Wang W, et al. Identification of an anti-gram-negative bacteria agent disrupting the interaction between LPS transporters LptA and LptC[J]. Int J Antimicrob Agents, 2019, 53: 442–448. DOI:10.1016/j.ijantimicag.2018.11.016 |
| [18] | Srinivas N, Jetter P, Ueberbacher BJ, et al. Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa[J]. Science, 2010, 327: 1010–1013. DOI:10.1126/science.1182749 |
| [19] | Werneburg M, Zerbe K, Juhas M, et al. Inhibition of lipopolysaccharide transport to the outer membrane in Pseudomonas aeruginosa by peptidomimetic antibiotics[J]. ChemBioChem, 2012, 13: 1767–1775. DOI:10.1002/cbic.201200276 |
| [20] | Takayama K, Qureshi N, Mascagni P, et al. Fatty acyl derivatives of glucosamine 1-phosphate in Escherichia coli and their relation to lipid A. Complete structure of a diacyl GlcN-1-P found in a phosphatidylglycerol-deficient mutant[J]. J Biol Chem, 1983, 258: 7379–7385. |
| [21] | Anderson MS, Bulawa CE, Raetz CR. The biosynthesis of gram-negative endotoxin. Formation of lipid A precursors from UDP-GlcNAc in extracts of Escherichia coli[J]. J Biol Chem, 1985, 260: 15536–15541. |
| [22] | Crowell DN, Anderson MS, Raetz CR. Molecular cloning of the genes for lipid A disaccharide synthase and UDP-N-acetylglucosamine acyltransferase in Escherichia coli[J]. J Bacteriol, 1986, 168: 152–159. DOI:10.1128/jb.168.1.152-159.1986 |
| [23] | Tomaras AP, McPherson CJ, Kuhn M, et al. LpxC inhibitors as new antibacterial agents and tools for studying regulation of lipid A biosynthesis in gram-negative pathogens[J]. mBio, 2014, 5: 1–13. DOI:10.3391/mbi.2014.5.1.01 |
| [24] | Barsotti RJ, Macy NE, Aubart MA, et al. Impact resistant transparent thermoplastic compositions: WO, 2014062601 [P]. 2014-04-24. |
| [25] | Titecat M, Liang X, Lee CJ, et al. High susceptibility of MDR and XDR gram-negative pathogens to biphenyl-diacetylene-based difluoromethyl-allo-threonyl-hydroxamate LpxC inhibitors[J]. J Antimicrob Chemother, 2016, 71: 2874–2882. DOI:10.1093/jac/dkw210 |
| [26] | Zhou P, Zhao J. Structure, inhibition, and regulation of essential lipid A enzymes[J]. Biochim Biophys Acta, 2017, 1862: 1424–1438. DOI:10.1016/j.bbalip.2016.11.014 |
| [27] | Montgomery JI, Brown MF, Reilly U, et al. Pyridone methylsulfone hydroxamate LpxC inhibitors for the treatment of serious gram-negative infections[J]. J Med Chem, 2012, 55: 1662–1670. DOI:10.1021/jm2014875 |
| [28] | Kalinin DV, Holl R. LpxC inhibitors: a patent review (2010-2016)[J]. Expert Opin Ther Pat, 2017, 27: 1227–1250. DOI:10.1080/13543776.2017.1360282 |
| [29] | Desroy N, Moreau F, Briet S, et al. Towards gram-negative antivirulence drugs: new inhibitors of HldE kinase[J]. Bioorg Med Chem, 2009, 17: 1276–1289. DOI:10.1016/j.bmc.2008.12.021 |
| [30] | Moreau F, Desroy N, Genevard JM, et al. Discovery of new gram-negative antivirulence drugs: structure and properties of novel E. coli WaaC inhibitors[J]. Bioorg Med Chem Lett, 2008, 18: 4022–4026. DOI:10.1016/j.bmcl.2008.05.117 |
| [31] | Desroy N, Denis A, Oliveira C, et al. Novel HldE-K inhibitors leading to attenuated gram negative bacterial virulence[J]. J Med Chem, 2013, 56: 1418–1430. DOI:10.1021/jm301499r |
| [32] | Champoux JJ. DNA topoisomerases: structure, function, and mechanism[J]. Annu Rev Biochem, 2001, 70: 369–413. DOI:10.1146/annurev.biochem.70.1.369 |
| [33] | Schoeffler AJ, Berger JM. DNA topoisomerases: harnessing and constraining energy to govern chromosome topology[J]. Q Rev Biophys, 2008, 41: 41–101. DOI:10.1017/S003358350800468X |
| [34] | Tari LW, Li X, Trzoss M, et al. Tricyclic GyrB/ParE (TriBE) inhibitors: a new class of broad-spectrum dual-targeting antibacterial agents[J]. PLoS One, 2013, 8: e84409. DOI:10.1371/journal.pone.0084409 |
| [35] | de Souza Mendes C, de Souza Antunes AM. Pipeline of known chemical classes of antibiotics[J]. Antibiotics (Basel), 2013, 2: 500–534. DOI:10.3390/antibiotics2040500 |
| [36] | Azam MA, Thathan J, Jubie S. Dual targeting DNA gyrase B (GyrB) and topoisomerse Ⅳ (ParE) inhibitors: a review[J]. Bioorg Chem, 2015, 62: 41–63. DOI:10.1016/j.bioorg.2015.07.004 |
| [37] | Morrow BJ, He W, Amsler KM, et al. In vitro antibacterial activities of JNJ-Q2, a new broad-spectrum fluoroquinolone[J]. Antimicrob Agents Chemother, 2010, 54: 1955–1964. DOI:10.1128/AAC.01374-09 |
| [38] | Chu DT, Fernandes PB, Claiborne AK, et al. Structure-activity relationships in quinolone antibacterials: design, synthesis and biological activities of novel isothiazoloquinolones[J]. Drugs Exp Clin Res, 1988, 14: 379–383. |
| [39] | Chu DT, Lico IM, Claiborne AK, et al. Structure-activity relationship of quinolone antibacterial agents: the effects of C-2 substitution[J]. Drugs Exp Clin Res, 1990, 16: 215–224. |
| [40] | Pucci MJ, Podos SD, Thanassi JA, et al. In vitro and in vivo profiles of ACH-702, an isothiazoloquinolone, against bacterial pathogens[J]. Antimicrob Agents Chemother, 2011, 55: 2860–2871. DOI:10.1128/AAC.01666-10 |
| [41] | Bax BD, Chan PF, Eggleston DS, et al. Type ⅡA topoisomerase inhibition by a new class of antibacterial agents[J]. Nature, 2010, 466: 935–940. DOI:10.1038/nature09197 |
| [42] | Dougherty TJ, Nayar A, Newman JV, et al. NBTI 5463 is a novel bacterial type Ⅱ topoisomerase inhibitor with activity against gram-negative bacteria and in vivo efficacy[J]. Antimicrob Agents Chemother, 2014, 58: 2657–2664. DOI:10.1128/AAC.02778-13 |
| [43] | Basarab GS, Doig P, Galullo V, et al. Discovery of novel DNA gyrase inhibiting spiropyrimidinetriones: benzisoxazole fusion with N-linked oxazolidinone substituents leading to a clinical candidate (ETX0914)[J]. J Med Chem, 2015, 58: 6264–6282. DOI:10.1021/acs.jmedchem.5b00863 |
| [44] | Basarab GS, Kern GH, McNulty J, et al. Responding to the challenge of untreatable gonorrhea: ETX0914, a first-in-class agent with a distinct mechanism-of-action against bacterial Type Ⅱ topoisomerases[J]. Sci Rep, 2015, 5: 11827. DOI:10.1038/srep11827 |
| [45] | Haranahalli K, Tong S, Ojima I. Recent advances in the discovery and development of antibacterial agents targeting the cell-division protein FtsZ[J]. Bioorg Med Chem, 2016, 24: 6354–6369. DOI:10.1016/j.bmc.2016.05.003 |
| [46] | Ma S, Ma S. The development of FtsZ inhibitors as potential antibacterial agents[J]. ChemMedChem, 2012, 7: 1161–1172. DOI:10.1002/cmdc.201200156 |
| [47] | Hogan AM, Scoffone VC, Makarov V, et al. Competitive fitness of essential gene knockdowns reveals a broad-spectrum antibacterial inhibitor of the cell division protein FtsZ[J]. Antimicrob Agents Chemother, 2018, 62: e01231–18. |
| [48] | Sun N, Ban L, Li M, et al. Probing the benzofuroquinolinium derivative as a potent antibacterial agent through the inhibition of FtsZ activity[J]. J Pharmacol Sci, 2018, 138: 83–85. DOI:10.1016/j.jphs.2018.09.001 |
| [49] | Bush K, Bradford PA. Beta-lactams and beta-lactamase inhibitors: an overview[J]. Cold Spring Harb Perspect Med, 2016, 6: a025247. DOI:10.1101/cshperspect.a025247 |
| [50] | Pattanaik P, Bethel CR, Hujer AM, et al. Strategic design of an effective beta-lactamase inhibitor: LN-1-255, a 6-alkylidene-2'-substituted penicillin sulfone[J]. J Biol Chem, 2009, 284: 945–953. DOI:10.1074/jbc.M806833200 |
| [51] | Vázquez-Ucha JC, Maneiro M, Martínez-Guitián M, et al. Activity of the β-lactamase inhibitor LN-1-255 against carbapenem-hydrolyzing class D β-lactamases from Acinetobacter baumannii[J]. Antimicrob Agents Chemother, 2017, 61: e01172–17. |
| [52] | Docquier JD, Mangani S. An update on beta-lactamase inhibitor discovery and development[J]. Drug Resist Updat, 2018, 36: 13–29. DOI:10.1016/j.drup.2017.11.002 |
| [53] | Zhanel GG, Lawrence CK, Adam H, et al. Imipenem-relebactam and meropenem-vaborbactam: two novel carbapenem-beta-lactamase inhibitor combinations[J]. Drugs, 2018, 78: 65–98. DOI:10.1007/s40265-017-0851-9 |
| [54] | McCarthy MW, Walsh TJ. Meropenem/vaborbactam fixed combination for the treatment of patients with complicated urinary tract infections[J]. Drugs Today (Barc), 2017, 53: 521–530. DOI:10.1358/dot.2017.53.10.2721815 |
| [55] | Lee YR, Baker NT. Meropenem-vaborbactam: a carbapenem and beta-lactamase inhibitor with activity against carbapenem-resistant Enterobacteriaceae[J]. Eur J Clin Microbiol Infect Dis, 2018, 37: 1411–1419. DOI:10.1007/s10096-018-3260-4 |
| [56] | Font H, Forner DF. American chemical society-255th national meeting and exhibition[J]. Drugs of the Future, 2018, 43: 365–373. DOI:10.1358/dof.2018.043.05.2808572 |
| [57] | Burns CJ, Daigle D, Liu B, et al. Beta-lactamase inhibitors: WO, 2014089365[P]. 2014-06-12. |
| [58] | Rao CVS, De Waelheyns E, Economou A, et al. Antibiotic targeting of the bacterial secretory pathway[J]. Biochim Biophys Acta, 2014, 1843: 1762–1783. DOI:10.1016/j.bbamcr.2014.02.004 |
| [59] | Smith PA, Koehler MFT, Girgis HS, et al. Optimized arylomycins are a new class of gram-negative antibiotics[J]. Nature, 2018, 561: 189–194. DOI:10.1038/s41586-018-0483-6 |
| [60] | Chu BC, Garcia-Herrero A, Johanson TH, et al. Siderophore uptake in bacteria and the battle for iron with the host; a bird's eye view[J]. BioMetals, 2010, 23: 601–611. DOI:10.1007/s10534-010-9361-x |
| [61] | Ji C, Juarez-Hernandez RE, Miller MJ. Exploiting bacterial iron acquisition: siderophore conjugates[J]. Future Med Chem, 2012, 4: 297–313. DOI:10.4155/fmc.11.191 |
| [62] | Mollmann U, Heinisch L, Bauernfeind A, et al. Siderophores as drug delivery agents: application of the "Trojan Horse" strategy[J]. BioMetals, 2009, 22: 615–624. DOI:10.1007/s10534-009-9219-2 |
| [63] | Isler B, Doi Y, Bonomo RA, et al. New treatment options against carbapenem-resistant Acinetobacter baumannii infections[J]. Antimicrob Agents Chemother, 2019, 63: e01110–18. |
| [64] | Page MG, Heim J. New molecules from old classes: revisiting the development of beta-lactams[J]. IDrugs, 2009, 12: 561–565. |
| [65] | Page MG, Dantier C, Desarbre E. In vitro properties of BAL30072, a novel siderophore sulfactam with activity against multiresistant gram-negative bacilli[J]. Antimicrob Agents Chemother, 2010, 54: 2291–2302. DOI:10.1128/AAC.01525-09 |
| [66] | Bush K. Investigational agents for the treatment of gram-negative bacterial infections: a reality check[J]. ACS Infect Dis, 2015, 1: 509–511. DOI:10.1021/acsinfecdis.5b00100 |
| [67] | Tan L, Tao Y, Wang T, et al. Discovery of novel pyridone-conjugated monosulfactams as potent and broad-spectrum antibiotics for multidrug-resistant gram-negative infections[J]. J Med Chem, 2017, 60: 2669–2684. DOI:10.1021/acs.jmedchem.6b01261 |