 2019, Vol. 54
2019, Vol. 54

编者按
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有 自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多维性质。本 栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
滴眼剂多佐胺的研制有如下的特点, 一是将研发目标由以往的口服全身用药变为眼睛局部滴入, 要求活性和组 织选择性以及溶解性和膜透入性等同时并举; 另一是对于生化机制和酶的三维结构以及抑制剂的作用机制认知明 确, 因而是在比较“透明”的环境中展开的。多佐胺作为基于酶蛋白结构设计成功的较早范例, 结构生物学、生化实 验和药物化学的构效关系等组合互补, 为调节微观结构和宏观性质的融合共处, 把构建结构中安排手性方向和基团 尺寸都做到了精雕细刻。
青光眼是指眼内压间断或持续升高的一种眼病, 持续的高眼压给眼球各部分组织和视功能带来损害, 如不及时治疗, 视野可以全部丧失而至失明。既有的降低眼压药物有胆碱能抑制剂、β肾上腺能阻滞剂、α肾上腺能激动剂、前列腺素和碳酸酐酶抑制剂等。碳酸酐酶抑制剂由于阻断碳酸氢根的生成, 减少眼液的产生而降眼压效果。
2 研发背景 2.1 眼内的碳酸酐酶碳酸酐酶在人体内有多种同工酶, 无处不在地分布于体内各种组织和器官中, 其功能是催化二氧化碳与水反应生成碳酸氢根(HCO3-), 将氧化代谢生成的CO2转化成调节内环境的pH因素。生成过程如图 1所示。不同亚型的碳酸酐酶作为药物靶标, 可用来治疗水肿、肥胖、骨质疏松和青光眼等疾病。眼部碳酸氢根的增加引起泪液增多, 使眼压增高, 成为青光眼的主要原因与征候。

|
Figure 1 Process for bicarbonate anion formation from carbon dioxide catalyzed by carbonic anhydrase |
治疗青光眼一直是口服全身用药, 由于碳酸酐酶在体内无处不在, 这会导致脱靶作用而引起不良反应。因而, 新的研发目标是局部滴眼抑制眼内的碳酸酐酶, 以减少不良反应。为制成滴眼剂需要药物有足够的水溶解性, 成均相的溶液; 但同时又得有一定亲脂性, 以穿越角膜, 进入眼内部以抑制靶标, 所以药物分子的亲水性和亲脂性的平衡是必要的。
2.2 碳酸酐酶的晶体结构碳酸酐酶的晶体结构早在1972年就已经X-射线获得解析, 并不断深化了对结构与功能的认识。碳酸酐酶是含有金属辅基的酶, 锌离子在活性中心的深部15Å 处, 位于疏水的裂隙边沿与数个组氨酸残基和水分子配位结合, 结构水分子参与了反应过程, His64咪唑环的亲核取代在催化过程起关键作用, 图 2是活性中心的结构配制。含有磺酰氨基团的抑制剂与锌离子发生配位结合, 竞争了水分子(后为羟基)的配位结合, 阻止了His64的正常功能。这些结构与功能对于分子设计起到指导作用。

|
Figure 2 Diagram of X-ray crystallography of carbonic anhydrase Ⅱ |
评价化合物活性分3个层次:体外(in vitro)、半体内(ex vivo)和体内(in vivo)。体外评价所用的酶来自人睫状突Ⅱ型碳酸酐酶的(HCA-Ⅱ), 用标准方法测定化合物的IC50, 同时测定化合物与HCA-Ⅱ的结合能力Ki值, 结果表明IC50与Ki值有平行关系。
半体内方法是评价化合物以足够浓度进入虹膜-睫状体而产生的药理效应为标准。为了降低眼球内压(IOP), 眼内的碳酸酐酶基本上被完全抑制, 为此, 局部滴入的受试液既能透入眼组织内, 也要抑制酶活性。用0.5%浓度的受试液滴入1 h后, 取家兔的虹膜-睫状体, 测定匀浆中碳酸酐酶受抑制的百分率。应当指出, 这种半体内方法并不能代表化合物体内抑制碳酸酐酶Ⅱ (CA-Ⅱ)的实际状态。
体内方法是用家兔的高眼压的α糜蛋白酶模型评价化合物的药理作用。局部滴入0.5%受试液1滴(50 μL), 在0.5~5 h的6个时间节点上测定眼压的降低值, 6只家兔的最大降低值的均值表示化合物的体内活性。
3.1 先导物——并环的噻吩磺酰胺项目在开始前临床用于治疗青光眼的药物是乙酰唑胺(1, acetazolamide)和伊索唑胺(2, ethoxzolamide), 两个药物都含有磺酰氨片段, 氮原子与辅基锌离子发生配位结合。由于溶解度低(< 1%), 难以制成均相滴眼剂, 口服用药产生不良反应。
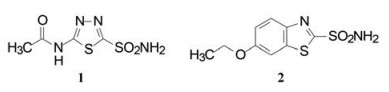
|
默克公司用溶解性大于1%的噻吩磺酰胺作为起始物, 合成了两个系列的化合物, 测定了化合物的溶解性和分配系数及对CA-Ⅱ的抑制活性(IC50)和结合常数(Ki), 列于表 1。
| Table 1 Physico-chemical property and activity of cyclosulfonylothiophene compounds |
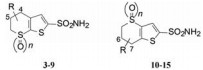
表 1中的化合物包含了两类骨架:噻吩并[2, 3-b]硫代吡喃(3~9)和噻吩并[3, 2-b]硫代吡喃(10~15)。结果表明, 两类骨架的化合物都有活性, 总的趋势是噻吩并[2, 3-b]硫代吡喃类的活性略高, 因此以后的结构优化都集中在这种骨架上。分析这些数据可获得如下构效关系: ①亚砜化合物(7和14)的活性显著低于硫醚和砜类化合物; ② 4位或7位的氧化状态对活性有影响, 酮基(3和13)的活性高于相应的羟基化合物(4和12)一倍; ③羟基的位置对活性影响不大, 例如4-OH和5-OH化合物(4和9、12和15)的活性基本相同; ④化合物5拆分后S构型为优映体, R为劣映体。
对表 1中的消旋化合物的活性与物化性质作多重相关分析, 得到如下的QSAR方程:
| $ \begin{array}{l} {\rm{log}}\left( {1/{\rm{I}}{{\rm{C}}_{50}}} \right) = \left( {0.61 \pm 0.17} \right){\rm{log}}P - \left( {0.49 \pm 0.22} \right){\rm{p}}{K_{\rm{a}}}\\ \;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\; + 3.926 \end{array} $ |
n = 13, r = 0.775, s = 0.469, F2, 10 = 7.52
式中log 1/IC50是活性的量度, 数值越大活性越强。log P是脂水分配系数的对数, 正值越大, 亲脂性越高。pKa是化合物的酸碱性量度, 数值越大碱性越强。n为参与回归分析的化合物样本数, r为方程的相关系数, s为标准偏差, F为检验方程置信度。
方程中的log P项系数为0.61, 表示化合物的亲脂性增加有利于活性的提高, 当提高分配系数一个对数单位活性增高4倍, 所以, 在一定范围内增加化合物的亲脂性有利于提高活性, 但不能无限制增高, 因为活性与亲脂性往往呈抛物线关系。pKa项的系数为负值, 提示降低pKa促进活性提高(Ponticello GS, Freedman MB, Habecker CN, et al. Thienothiopyran-2-sulfonamides: a novel class of water-soluble carbonic anhydrase inhibitors. J Med Chem, 1987, 30: 591-597)。然而QSAR方程中pKa项的系数只是在7.9~9.9的狭窄范围内推断的, 实际上引入碱性氮元素是有利于活性的。
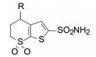
3.2 噻吩并硫代吡喃环上引入碱性侧链多亏没有在上述QSAR的pKa项“误导”下分子中引入酸性基团, 反而连接了碱性, 虽然在发表的原始文献中没有叙述设计的依据, 但估计是为了提高化合物的水溶性加入了碱性氮原子。同时也考察变换亲脂性对透过角膜进入眼内的影响。合成的化合物列入表 2 (Baldwin JJ, Ponticellot GS, Anderson PS, et al. Thienothiopyran-2-sulfonamides: novel topically active carbonic anhydrase inhibitors for the treatment of glaucoma. J Med Chem, 1989, 32: 2510-2513)。
| Table 2 Structures and inhibition of N-containing compounds for carbonic anhydrase |
 OH
OH OH
OH NHCH2CH3
NHCH2CH3
 NHCH2CH3
NHCH2CH3
 NHCH2CH(CH3)2
NHCH2CH(CH3)2
 NHCH2CH(CH3)2
NHCH2CH(CH3)2
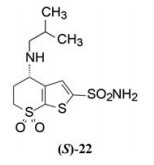
分析表 2的数据可提示如下构效关系: ①胺基系列化合物IC50和Ki的数据具有平行和对应性; ②羟基被胺基替换保持或提高了活性, 但须保持伯胺或仲胺结构, 因为化合物19为二乙胺基的叔胺活性显著减弱; ③化合物18和22活性最强, 经拆分后的活性评价表明S构型为优映体, 尤其是22S的活性强于22R上百倍, 该立体专属性差异的原因在下一节讨论。
3.3 化合物的半体内和体内活性用家兔局部滴眼的半体内和体内方法评价胺类化合物的数据列于表 3。半体内方法测定受试物对眼部碳酸酐酶的抑制率和整体实验的降眼压作用, 反映了局部滴入受试物的穿越眼组织的能力、抑制碳酸酐酶的活性和降低眼压的效果。综合评价结果表明化合物22优于其他化合物, 尤其是拆分的S-异构体, 完全抑制了酶活性, 而且降低眼压的持续时间长。
| Table 3 Biological properties of 5, 6-dihydro-4H-thieno[2, 3-b]thiopyran-2-sulfonamide 7, 7-dioxide derivatives. *One hour after the topical administration (one drop, 50 μL) of vehicle or test compound at 0.5%. The iris-ciliary body was excised from albino rabbits and the tissue homogenized. Carbonic anhydrase activity was determined by using a pH stat assay. **Ocular hypertension was induced in the right eye of albino rabbits by the intraocular injection of a-chymotrypsin.13 Test compound at 0.5% was instilled (one drop, 50 μL) and the IOP of six rabbits measured just before (t0) and at 0.5, 1, 2, 3, 4, and 5 h after treatment. Results are expressed as the maximum fall (mean ± SE) in IOP (mmHg) from the t0 value |
 NHCH2CH(CH3)2
NHCH2CH(CH3)2
 NHCH2CH(CH3)2
NHCH2CH(CH3)2
S-22是里程碑化合物, 代号为MK-417进行了深入研究。
3.4 化合物22的结合模式为了探索化合物22的结合模式, 分析S-22高于R-22的抑制活性100倍而成为优映体的原因, 用X-射线衍射分析了S-22和R-22与CA-Ⅱ晶体结构。二者相同和不同的要点如下: ① S-22与R-22的噻吩并硫代吡喃骨架在酶活性中心的取向与定位是相同的; ②磺酰胺基的氮负离子与锌形成配位键, 成为锌的第4个变形的配体; ③ CA-Ⅱ活性中心的重要残基His64 (用于拔除配位水分子的质子, 见图 1)位置移动, 与标准位置相比, 咪唑环移动3.2 Å ; ④在与酶结合之前, S-22和R-22的异丁胺基侧链在硫代吡喃环上采取假平展键, 结合后S-22的取向变为假直立键, 以适配于疏水腔内, 此时并合的氨磺酰噻吩的N-S-C-S两面角由(量子化学ab initio计算) 90°扭变为144°, 呈trans构象; R-22虽然结合前后的异丁胺基侧链都采取假平展键, 但为适配于疏水腔的结合, 将N-S-C-S两面角由90°扭变为158°, 取gauche构象, 两个对应体的两面角差值14°需要能量补偿, 加之S-22的硫代吡喃环上的砜基与酶的Gln92氢键距离2.7 Å ,而R-22为3.1 Å ,总体的结合状态表明S-22呈优势结合。图 3是S-22和R-22结合状态的分子构象示意图。
|

|
Figure 3 a. Conformational diagram of S-22 at binding condition; b. Conformational diagram of R-22 at binding condition |
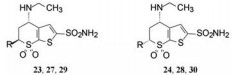
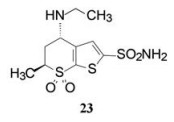
为了去除S-22与碳酸酐酶的结合过程中胺基侧链的构象变化而发生的能量损耗, 在硫代吡喃环的6位引入甲基, 形成新的手性中心, 使甲基的引入“逼迫”胺基侧链采取能量适宜的构象, 因而需要考察两对差向异构体的优势结合与高活性的关联性。同时, 为降低由于引入甲基而增加的分子亲脂性, 将异丁胺基变为乙胺基, 合成并分离/拆分了4个异构体trans-S, S-23、cis-S, R-23、trans-R, R-23、cis-R, S-23, 分别测定了抑酶活性, 并制备和解析了复合物单晶的结构, 表 4列出了4个异构体的数据。
| Table 4 Structure, activity and binding energy of compound 23 in different isomers |
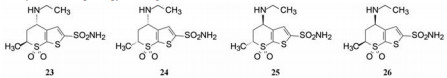
比较一对trans对应异构体S, S (23)和R, R (25)的活性, 显示S, S构型的活性显著强于R, R, 另一对cis对映体S, R (24)活性强于R, S (26), 显示出以4-乙胺基为S构型的化合物活性占优。四个化合物的晶体结构的N-S-C-S两面角与结合前真空态(90°)的差值表明, S, S两面角的偏离最少, 而R, R的偏离值最大, 与活性的变化呈正相关。此外, 活性也与构象能的损失相一致, 只1.5 kcal·mol-1的能量就导致结合活性差大约20倍。晶体的三维结构也揭示了该两面角的不同所诱导的酶重要残基His64发生迁移的程度也不同, His64的位置变迁对于碳酸酐酶催化反应是非常重要的。
4.2 优化6位烷基为了优化6位烷基, 固定4位S-乙胺基不变, 合成的6S-和6R-烷基的化合物列于表 5中。6位烷基为S构型的化合物活性比相应的R构型活性高。6S-系列的甲基、乙基和丙基化合物的活性都很高, 提示6位烷基结合的疏水腔有足够的空间。分子对接表明, 该疏水腔可容纳到丙基的体积而不会影响2位磺酰氨基与锌离子的结合, 也不干扰4位乙胺基对His64的影响。虽然丙基活性略高于甲基, 但为保障化合物的水溶解性, 优选了甲基(Greer J, Erickson JW, Baldwin JJ, et al. Application of the three-dimensional structures of protein target molecules in structure-based drug design. J Med Chem, 1994, 37: 1035-1054)。
| Table 5 Activity of varied 6-alkyl compounds |
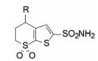
优化至此, 化合物23显示有强效抑制碳酸酐酶Ⅱ的活性和良好的物理化学性质, 水溶解度0.699 mg·mL-1, log P -0.5, pKa 8.18, 极性表面积106.33 Å2。虽然只用作外用的滴眼剂, 但也系统地研究了药代动力学性质和安全性, 最终确定化合物23为候选化合物, 定名为多佐胺(dorzolamide), 以2%水溶液外用滴眼剂进行临床研究, 表明对青光眼患者可降低眼高压, 由于不经口服用药, 避免了许多不良反应, 美国FDA于1995年批准上市。
|
X-射线分析多佐胺与碳酸酐酶Ⅱ的复合物晶体结构表明(图 4), 多佐胺的2位磺酰氨取代了正常状态的羟基负离子与锌配位结合, 氮原子还与Thr199的羟基, 磺酰基的氧原子与Gln92分别发生氢键结合。4位S-乙胺基的重要性表现在占据了疏水空间, 迫使His64的咪唑侧链移动, 离开了正常的催化状态, 同时也“挤掉”一个结构水分子成自由的无序态, 而无抑制剂或4S-氨基或4S-甲胺基化合物仍保持该水分子的有序态, 因而这部分结构的有利熵效应成为多佐胺高抑制活性的结构所在。6S-甲基所占据的平展键, 对于4S-乙胺基采取假直立构象的取向起到了稳定作用。

|
Figure 4 X-ray chrystallography of dorzolamide-CA Ⅱ complex (PDB code 1CIL) |