 2019, Vol. 54
2019, Vol. 54

编者按
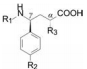
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多维性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
2015年7月美国FDA批准诺华的复方制剂Entresto上市, 治疗慢性心力衰竭, 由于能够显著地降低心衰患者的死亡率, 被认为是25年来心衰治疗药的重大突破。Entresto是由沙库必曲和缬沙坦组成。沙库必曲作为新分子实体, 是以复合制剂被批准面市的, 两个主成分针对不同的靶标各司其责和药理作用的互补性, 使得Entresto问世具有划时代性。在剂量设计和分子结构上也很有特色:两个成分的治疗剂量恰好是等摩尔比量; 与钠离子形成配位键结合以及分子内和分子间的氢键网络, 构成了稳定的超分子结合的共晶结构。本文简述了研制沙库必曲的药物化学过程。
2015年7月美国FDA批准诺华的复方制剂Entresto上市, 治疗慢性心力衰竭, 这个复方制剂是以多种方式作用于心脏的神经内分泌系统, 治疗效果与标准疗法比较, 显著地降低心衰患者的死亡率, 被认为是25年来心衰治疗药的重大突破。Entresto是由沙库必曲(1, sacubitril, 49 mg)和缬沙坦(2, valsartan, 51 mg)组成。沙库必曲的作用靶标是抑制一种中性内切酶(neprilysin), 中性内切酶是降解血浆中心房利钠肽(artrial natriuretic factor, ANF)的水解酶, 因而沙库必曲使内源性ANF肽维持在一定的水平, 保护了心脏的神经内分泌系统, 有利于心衰患者; 缬沙坦是血管紧张素Ⅰ受体阻断剂, 抑制AngⅡ的升压作用, 阻断患者过于活跃的肾素-血管紧张素-醛固酮系统(RAAS)。复方制剂Entresto中的沙库必曲是以新分子实体(NME)首次上市的, 本文简述其研制的药物化学轨迹。
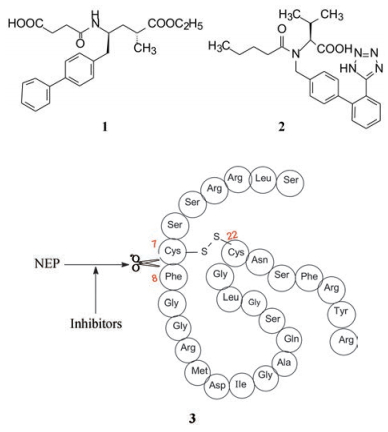
2 心房钠肽——舒张血管的内源肽心房钠肽(3, ANF, 又称心房钠尿因子)是由心肌细胞产生的28肽, 分泌到外周血中, 产生排钠、利尿和舒张血管效应, 调整血压、体液容量和电解质平衡。在功能上ANF虽与肾素-血管紧张素系统(RAS)相反, 但二者是完全独立的系统, 所以构成了Entresto作为针对不同靶标制成复方药物的基础。
静脉滴注ANF可产生持续性降压和排钠作用, 但半衰期短, 不能口服应用。克服这种局限性的方法是阻断水解ANF的中性内切肽酶(NEP)功能。NEP切断ANF的Cys7-Phe8肽键而失活, 体内外实验表明NEP酶抑制剂与滴注低剂量ANF可产生同样的降压效果。本文阐述的就是NEP酶抑制剂的研制过程。
|
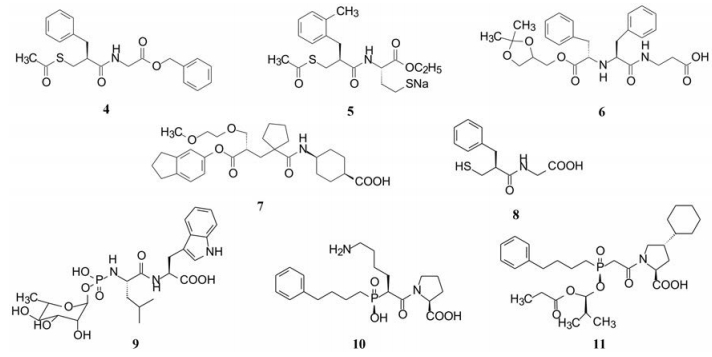
1993年Ciba-Geigy公司(今为诺华)在开始研发时已经有数个候选化合物处于临床研究阶段, 例如sinorphan (4)、SCH-42495 (5)、SCH-34826 (6)和UK-69578 (7)等, 但临床治疗降压效果一般, 这些候选物均未成功上市, 原因可能是药效作用不强或药代不合理。分析这些NEP抑制剂的结构, 都存在有羧基(4和5是羧酸的前药)或硫醚基, 这些基团的功能是与NEP的Zn辅基配位结合。同时在相隔一定距离存在疏水性片段, 可能与酶的疏水腔发生疏水-疏水相互作用。
4 活性评价为了评价化合物对NEP抑制活性, 用合成的戊二酰-丙氨酰-丙氨酰-苯丙氨酰-2-萘胺(GAAP)为底物, 通过测定剩余GAAP的浓度, 作为评价受试化合物抑制NEP的能力, 用已知抑制剂thiorphan (8, IC50 = 4.8 nmol·L-1)或phosphoramidon (9, IC50 = 27.1 nmol·L-1)作为阳性对照药。
5 可口服吸收但不进入细胞内的磷(膦)酸酯类前药肾脏和血管中分布有较多的NEP酶, 对许多内源性肽(例如脑啡肽、ANF等)有水解失活作用, 通过与活性中心的辅基锌离子螯合水解特定的肽键而迅速失活。ANF有较低的分布容积, 提示抑制剂最好存在于细胞外, 减少进入细胞发生脱靶作用。
磷(膦)酸基易于同锌离子发生螯合作用, 但化合物带有游离的负电荷不易被机体吸收, 血管紧张素转化酶(ACE)也是经锌离子介导的肽酶, ACE抑制剂与锌离子螯合的基团有巯基、羧基或磷酸基, 例如上市的ACE抑制剂西那普利(10, ceranapril)和福辛普利(11, fosinopril)是以有机膦酸酯代替羧酸酯而延长了作用时间, 本项目因而设计了含有可与锌离子螯合的含磷(膦)酸基的化合物。
6 探索含磷酸基的基本骨架 6.1 初始的α-磷酸基苯丙酰胺骨架化学结构设计的构想是:分子中含有氨基酸片段, 有磷酸基团与辅基Zn结合, 亲脂链与酶的疏水腔发生疏水-疏水相互作用。基于这些因素的配置, 合成的有代表性的苯丙酰胺基丙酸类化合物列于表 1中。
| Table 1 Activity of phosphono-containing phenylpropionamido-propionic acid compounds |
表中数据提示, 只有化合物12呈现中等强度活性, 活性低于阳性对照7倍, 13~16的活性很差。原因可能是①磷(膦)酸基被两个较大疏水基团包夹, 位阻效应不利于同锌发生配位结合; ②疏水链不适宜的位置而未进入酶疏水腔; ③缺乏α-氨基, 不能提供氢键给体, 导致结合力弱。
6.2 引入α-氨基和调整链长为了使α-氨基具有氢键给体作用, 在氨基与膦酸基之间用CH2隔开, 以避免氨基与膦酰基直接相连的共轭效应而减弱氢键给体效应, 合成的有代表性化合物列于表 2。
| Table 2 Structures and activity of compounds with α-N-phosphonomethyl amino skeleton |
表 2的构效关系提示, ① α碳连接的苄基(化合物17)变换为联苯甲基(18), 体外抑制NEP活性提高600倍; ② α碳的S构型为优映体, R构型(19)没有活性, R, S-混旋体(20)仍有强活性, 说明抑制剂在这个位置需有足够显著的立体选择性以与疏水腔结合; ③磷酸基被不同程度的酯化, 体外活性显著减弱, 基团尺寸越大减弱的越多(例如化合物21~23), 提示该处存在位阻效应, 影响磷酸基与Zn发生的螯合作用(形成一齿或二齿螯合); ④变换羧基与α碳连接链的距离(加长或缩短如25~27)或间隔基上的取代(28和29)对活性影响不大, 说明该处的构效关系比较宽松。
|
化合物18不仅对NEP抑制活性高而且选择性强, 例如在20 μmol·L-1浓度下对血管紧张素转化酶、嗜热菌蛋白酶、内皮素转化酶和基质溶解因子等酶系都没有脱靶作用。
6.3 化合物18的药代动力学和前药设计清醒大鼠静脉注射10 mg·kg-1化合物18, 半体内方法在不同的时间间隔取血样, 测定GAAP量以评价血浆中游离18的浓度, 结果表明, 在给药后至少4 h内18仍有较高的血药浓度, 高于IC50值2~3数量级。
然而化合物18含有3个可离解的酸性基团, 难以穿越细胞膜, 所以大鼠灌胃的生物利用度低, 只有3%。为增进口服吸收, 合成了一系列可水解的酯性前药, 包括膦酸基或羧酸基的酸性基团部分或全部酯化, 发现化合物31大鼠灌胃30 mg·kg-1, 4 h后血浆中的31具有相当高的水平, 与活性代谢物18的IC50之比为225 (灌胃4 h后血浆中药物浓度超过IC50值200倍)。而羧基同时被乙酯化的比值为160, 提示全部酯化对吸收未必有利(也因此后述的沙库必曲中两个羧基只有一个被乙酯化)。31是个有希望的候选化合物(De Lombaert S, Erion MD, Tan J, et al. N-Phosphonomethyl dipeptides and their phosphonate prodrugs, a new generation of neutral endopeptidase (NEP, EC 3.4.24.1 1) inhibitors. J Med Chem, 1994, 37: 498-511)。
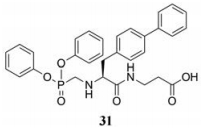
|
中性内切酶(NEP)高表达于中枢神经和肾脏, 可水解Met-脑啡肽, 通过剪切Gly3-Phe4肽键, 使脑啡肽的镇痛作用失活。NEP也可水解其他内源性肽如缓激肽、P物质和神经降压肽等, 通过研究抑制NEP对脑啡肽的水解作用, 旨在研发具有选择性的长效镇痛药物。所以, 评价这类化合物的体外抑制NEP活性是用Met-脑啡肽(Met-ENK)作底物。
7.2 连接氨基酸的2, 4-二苄基戊二酸化合物作为模拟二肽类分子, 合成了系列的氨基酸的2, 4-二苄基戊二酸化合物, 将天然的氨基酸或β氨基酸的1位羧基酰化, 得到的化合物及其抑制脑啡肽的活性列于表 3。
| Table 3 SAR of compounds with 2, 4-dibenzyl glutaric acid skeleton |
 (RS, RS)
(RS, RS) (S, S)
(S, S)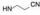 (S, S)
(S, S)
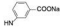 (RS, RS)
(RS, RS)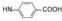 (S, S)
(S, S)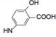 (S, S)
(S, S)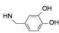 (S, S)
(S, S)分析表 3的结构与活性关系, 可得到如下信息: ①改变氨基酸的链长, 由甘氨酸(32)到β丙氨酸(34)到γ氨基丁酸(37)都具有高活性, 提示对应于酶的S2'位置可容纳一定长度的柔性链, C2~C4的活性没有显著变化。② S2'处羧基是重要的, 换成氰基(45)或羟基(46)活性降低至少100倍。③邻氨基苯甲酸(43)模拟构象限制的β-丙氨酸, 失去了活性, 说明羧基与氨基呈顺式构型不利于结合。邻氨基苯乙酸(44)模拟构象限制的γ-氨基丁酸, 仍有高活性, 提示该柔性键有利于形成活性构象。④连接两个苄基的手性碳以S, S-构型为优映体(34), 其对映体R, R的活性很低(35), 提示该处的疏水-疏水相互作用具有较强的立体选择性。⑤将两个苄基换成苯乙基, 或苄基的苯环换成环己甲基, 活性都显著降低(表 3未列出), 进一步提示两个苄基对于呈现活性的重要性。
7.3 反向酰胺化合物的活性上述化合物34有很强的体外抑制脑啡肽酶的活性, 进而设计合成了将酰胺键以相反的连接方式连接的化合物, 考察氢键给体的位置变化对活性的影响, 这样母核由原来的二苄基戊二酸变成二苄基氨基丁酸, 表 4列出了目标化合物的活性。
| Table 4 SAR of 2, 4-dibenzyl-4-amino-butyramide compounds |
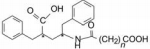
由表 4可以看出, 反向酰胺化合物仍然保持了抑制脑啡肽酶的活性, 其中54的活性略强于34, 而且手性碳的构型仍以S, S的活性最强(Ksander GM, Diefenbacher CG, Yuan AM, et al. Enkephalinase inhibitors. 1. 2, 4-Dibenzylglutaric acid derivatives. J Med Chem, 1989, 32: 2519-2526)。
8 联苯戊二酰胺化合物 8.1 2, 4位亲脂链不对称的化合物脑啡肽酶和neprilysin虽然都是内源性肽的水解酶, 但两个酶的抑制剂的选择性表现是不同的, 例如thiorphan (8)和phosphoramidon (9)对二者都有较强的活性(作为先导化合物这是个优点, 但作为药物就可能因为脱靶作用而呈现不良反应), 然而上节讨论的二苄基戊二酸和氨基丁酸化合物34和54虽然对脑啡肽酶有强效抑制活性, 但对neprilysin的活性很弱, IC50分别为1 200 nmol·L-1和4 000 nmol·L-1。
借鉴前述的氨基膦酸化合物的结构中的联苯甲基具有较强的活性贡献, 因而将该片段引入到戊二酸系列中, 以ANF为底物评价对酶的抑制活性。表 5列出了含有一个联苯甲基的戊二酸酰胺的化合物。
| 表 5 SAR of glutaramide compounds containing biphenylmethyl group |
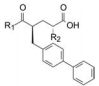
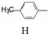
表 5中的化合物是固定联苯甲基不变, 变换R1的不同氨基酸和R2的基团, 分析构效关系如下: ① R1由β丙氨酸换成异丝氨酸, 活性提高大约3~5倍; ②固定联苯甲基和β丙氨酸不变, 变换R2基团成烷基、烷氧基、芳烷基或芳氧基, 对活性影响不显著, 但无取代的H活性降低4倍; ③手性碳原子的构型很重要, 如成对的化合物59与60、61与62的活性比较, 差向异构体几乎失去了活性。
8.2 2, 4位亲脂链相同的化合物对称性的戊二酰β丙氨酸的骨架上当2, 4位是二苄基时(化合物34), 抑制活性很弱, 但换成联苯甲基(67)或吡咯苯甲基(68)活性显著提高, 不过, 换成其他取代苯, 活性也较弱。
| Table 6 SAR of symmetric glutaramide compounds |
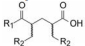

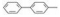
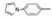




上一节合成的含有双苄基的γ胺基丁酸与氨基酸形成酰胺的化合物, 例如54的活性很强, IC50值为2 nmol·L-1, 设想若含有联苯基的不对称化合物会有较强的活性。根据表 5和表 6的构效关系, 可以演绎出α碳连接烷基或芳烷基对活性影响不大, 而γ位置的联苯甲基则是最佳的抑制基团, 因而设计的化合物是R1为丙二酰、丁二酰、丁酰和戊二酰基, R2为苯基, R3为甲基或甲氧基(小尺寸原则), 化合物列于表 7中。
| Table 7 Asymmetric amino-butyramide compounds |
表中数据显示, 活性最强的是化合物74, IC50值5 nmol·L-1, 4个立体异构体中以αR, γS活性最强, αS, γS (77)的活性其次, γ位为R构型的化合物(75和76)活性显著减弱。α位换成甲氧基(81)与甲基活性相当, 无取代的H (82)活性较弱。R1片段以丁二酰基活性最强, 增加一碳(戊二酰)或减少一碳(丙二酰)都使活性下降。
9 不同结构类型的体外活性比较以上不同结构类型的化合物对中性内切酶(NEP)的抑制活性分别用不同的肽类底物进行了评价, 即戊二酰-Ala-Ala-Phe-酰萘胺(GAAP)、Leu-脑啡肽(Leu ENK)和心房利钠肽(ANF), NEP对这3个底物的催化作用Kcat/Km分别为37、56、18。
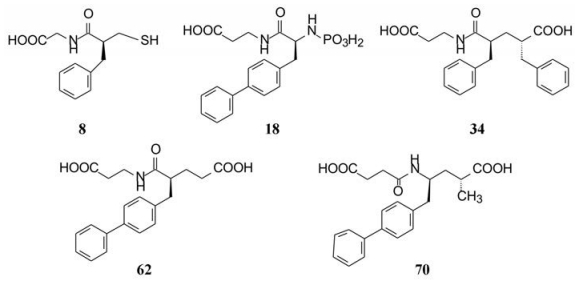
为了在不同结构类型中选择最佳的化合物, 挑选前述高活性的化合物作进一步比较, 这些化合物是8、18、34、66和74。
表 8的数据显示, 含巯基的thiorphan (8)、胺基膦酸化合物18和二羧酸类化合物74用3种不同底物测定对NEP的抑制活性(IC50值)是相似的, 虽然用Leu-脑啡肽的模型活性略偏高, 但这3个化合物的体外活性具有体内的预测性。然而化合物34和66在GAAP和Leu-ENK为底物测定的活性相差很大, 就难以预测体内活性, 这可能是由于34和66的动力学性质不同的缘故。
| Table 8 Inhibitory activities of high active compounds on NEP by various substrates measurement |
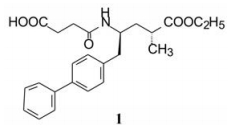
化合物74在体内外模型评价中显示较高抑制活性, 但由于结构中含有两个羧基, 极性过强不易吸收, 因而制成单酯性前药1, 以钠盐形式在大鼠、犬和猴评价药代动力学性质。化合物1以剂量30 mg·kg-1灌胃大鼠和猴或十二指肠给药麻醉犬, 测定血液中水解成原药74 (未与蛋白结合的游离形式)的浓度, 在1~6 h内生成的化合物74的浓度分别为0.64~0.05 μmol·L-1 (大鼠)、2.8~0.08 μmol·L-1 (犬)和8.51~0.21 μmol·L-1 (猴), 比体外测定对NEP抑制的IC50高1 500~38倍。活性化合物74在猴血浆中半衰期(t1/2)为4.6 h (Ksander GM, Ghai RD, de Jesus R, et al. Dicarboxylic acid dipeptide neutral endopeptidase inhibitors. J Med Chem, 1995, 38: 1689-1700)。
|
进而对化合物1用大鼠和犬进行体内ANF的影响和麻醉犬ANF诱导的排钠和利尿实验, 结果表明1有确定的药理作用。化合物1命名为沙库必曲(sacubitril)与血管紧张素受体拮抗剂缬沙坦(2)合用治疗心力衰竭患者, 经三期临床研究, 于2015年FDA批准沙库必曲和缬沙坦作为固定复方制剂上市, 商品名为Entresto (Voors AA, Dorhout B, van der Meer P. The potential role of valsartan + AHU377 (LCZ696) in the treatment of heart failure. Expert Opin Investig Drugs, 2013, 22: 1041-1047)。
作为固定剂量的复方制剂, 应用Entresto起始剂量是49/51 mg (沙库必曲/缬沙坦)每天2次。当患者耐受后, 2至4周后加倍Entresto剂量至目标维持剂量97/103 mg (沙库必曲/缬沙坦)每天2次。
|
Entresto并非是沙库必曲和缬沙坦混合物, 而是等摩尔量组成的共晶复合物。经X-射线衍射分析, 每个共晶体由6个沙库必曲负离子、6个缬沙坦负离子、18个钠离子(用的氢氧化钠量不是化学计量的)和15个水分子构成, 分子式为C288H330N36Na18O48·15H2O, 分子量为5 748.03, 为白色六边形片状结晶形粉末, 熔点138 ℃。复合物在固态和pH 5~7的水溶液是稳定的。
作为超分子的钠复合物, 18个钠离子与来自沙库必曲和缬沙坦的12个羧基和18个羰基的氧原子, 以及15个水分子中的13个氧原子形成配位结构, 如图 1所示。超分子还由于形成氢键网络而稳定化。缬沙坦的四唑环没有与钠形成离子键, 而是与沙库必曲的酰NH形成氢键(Feng L, Karpinski PH, Sutton P, et al. LCZ696: a dual-acting sodium supramolecular complex. Tetrahedron Lett, 2012, 53: 275-276)。

|
Figure 1 Crystallography of Entresto complex. Red ball stands for sodium ion, green is water molecule |
沙库必曲作为新分子实体, 是以复合制剂被批准面市的, 两个主成分的药理作用的各司其责并具有互补性, 使得Etresto在治疗心衰患者成为划时代的突破。在剂量设计和分子结构上也很有特色:两个成分的治疗剂量恰好是等分子比量; 与钠离子形成配位键结合以及分子内和分子间的氢键网络, 构成了稳定的超分子结合的共晶结构, 也因此获得知识产权保护, 克服了这两个单体的专利已经过期的局面。复方制剂有时并非是简单的物理混合物(Thipparaboina R, Kumar D, Chavan RB, et al. Multidrug co-crystals: towards the development of effective therapeutic. Drug Discov Today, 2016, 21: 481-490)。