 2018, Vol. 53
2018, Vol. 53

编者按
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多维性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
伏立诺他是作用于组蛋白去乙酰化酶的首创性抗肿瘤药物, 该药物的成功研制给人们以诸多启示。基础研究偶然观察到DMSO对白血病细胞的诱导分化作用, 激发出创新的热情, 在不知靶标和缺少机制认识的情况下, 硬是从最简单的分子, 大胆的假设和科学的求证, 最终“长成”了伏立诺他分子结构。当后来发现并验证了组蛋白去乙酰化酶为药物靶标后, 竟发现伏立诺他与酶活性部位呈高互补性的“严丝合缝”, 再一次证明通过生理表型的变化研发新药、药物化学和构效关系的研究, 仍是行之有效的方法。条条大路通罗马。摸着石头过河, 可以到达胜利的彼岸。
伏立诺他(1, vorinostat)是组蛋白去乙酰化酶(HDAC)抑制剂, 于2007年经美国FDA批准上市, 治疗皮肤T淋巴细胞癌。作为首创性药物的HDAC抑制剂, 从化学结构看伏立诺他很像是真菌的代谢产物曲古抑菌素A (2, trichostatin A)的简化物, 曲古抑菌素是具有抑制HDAC活性的天然产物, 1和2的结构具有相似性, 会以为伏立诺他是基于天然活性物质优化结构而成的药物, 实际上追踪研发过程, 构建伏立诺他的化学结构与曲古抑菌素A没有什么关系, 只是后来在确证其作用靶标时是个重要的参照物。伏立诺他的研制是源自于简单的分子二甲基亚砜(DMSO)。
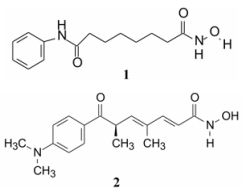
|
Friend等在研究小鼠红白血病细胞(MELC)转染的抑制作用中, 所用的细胞培养液中含有浓度为280 mmol·L-1的DMSO缓冲液, 发现有三分之二的癌细胞颜色变红, 提示DMSO可能刺激了MELC的细胞分化, 但不抑制细胞生长, 并由于提高了血红蛋白的合成, 生成正红细胞(normoblast), 减少了细胞的恶变(Friend C, Scher W, Holland JG, et al. Hemoglobin synthesis in murine virus-induced leukemic cells in vitro: stimulation of erythroid differentiation by dimethyl sulfoxide. Proc Natl Acad Sci USA, 1971, 68: 378-382)。这样一个偶然发现的极性小分子诱导MELC细胞的分化, 并且抑制白血病细胞的突变作用, 激励了Breslow等的深入研究, 并将DMSO作为研究抗白血病药物的苗头化合物。
3 苗头演化为先导化合物 3.1 评价活性的模型评价化合物对小鼠红白血病细胞的体外诱导分化活性, 采用了两种方法, 一是不同浓度的受试化合物与白血病细胞温孵, 5日后测定联苯胺被氧化的生色反应, 评价细胞中的血红蛋白(高铁离子)的百分含量, 颜色越深, 表明诱导分化作用越强; 另一方法是将化合物与每毫升含有1×107的细胞温孵, 然后将细胞消化液在415 nm波长下测定吸光度, 确定血红蛋白的含量。
3.2 极性分子结构的多样性Breslow等将DMSO扩展到其他类型的极性分子, 用病毒感染小鼠诱发的红白血病细胞评价它们诱导分化的活性, 发现N-甲基甲酰胺(3)、N, N-二甲基甲酰胺(4)、乙酰胺(5)、丙酰胺(6)、N-甲基乙酰胺(7)、N, N-二甲基乙酰胺(8)、哌啶酮(9)、1-甲基哌啶酮(10)、2-吡咯烷酮(11)、N-甲基吡咯烷酮(12)、吡啶-N-氧化物(13)和三乙烯乙二醇(14) (图 1)等都具有类似于DMSO诱导MELC分化的作用, 推论细胞在合成DNA的过程中这些极性化合物可能改变了DNA或DNA-蛋白复合物的构象, 导致细胞转染过程和红细胞分化程序的表达发生变化(Tanaka M, Levy J, Terada M, et al. Induction of erythroid differentiation in murine virus infected eythroleukemia cells by highly polar compounds. Proc Natl Acad Sci USA, 1975, 72: 1003-1006)。

|
Figure 1 The polar molecules possessing induction of differentiation and inhibition of erythroleukemia cells |
为了提高化合物的诱导分化活性, 研究者确定了如下的目标: ①提高化合物活性, 降低有效浓度; ②提高诱导分化率; ③消除或降低细胞毒作用。为此, 以N-甲基乙酰胺(7)为起始物, 设计合成7的二聚化合物, 希望将两个极性基团连接成一个分子, 增加与未知靶标相近位点的结合机会, 这在热力学上使结合处于有利的熵状态。为此, 以7的甲基为连接点, 通过不同数量的亚甲基(n = 2, 4~8), 变换连接基的长度, 分析构效关系, 优化结构。合成的化合物15~21及其活性列于表 1。
| Table 1 Activity of inducing differentiation for bisactetodiamides (15-20) and bispropiondiamides (21). *Cell growth inhibition |
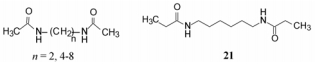
表 1数据显示, 最简单的双酰乙二胺15 (n = 2)的活性并不强于化合物7, 说明过于接近的功能基未能结合到重要的位点上, 无助于提高活性。当加长连接基n = 5~8 (17~20)时, 化合物活性显著增高, 在低浓度下也有较高的诱导分化率。虽然n = 7或8 (19, 20)的化合物在低浓度(3 mmol·L-1)下有较高的分化活性, 与n = 5或6在5 mmol·L-1的效应相同, 但19和20化合物已呈现毒性, 细胞的生长受到抑制。然而在5 mmol·L-1浓度下, 化合物17和18 (n = 5和6)诱导细胞的分化率最高, 而且不产生细胞毒作用。21是己二胺的双丙酰化合物, 虽然也有较高的诱导分化活性, 但在产生最高活性的7 mmol·L-1浓度下, 显著抑制细胞生长。化合物18为N, N-二乙酰六亚甲基二胺(简称为HMBA), 证明为在活性和毒性之间有显著区分的优选分子, 为里程碑式的化合物。
深入研究HMBA对细胞分化的行为, 表明在0.5~5 mmol·L-1内诱导分化作用呈现浓度依赖关系, 而且分化程度也呈时间依赖性, 但浓度增高则出现细胞毒作用(Reuben RC, Wife RL, Breslow R, et al. A new group of potent inducers of differentiation in murine erythroleukemia cells. Proc Nat Acad Sci USA, 1976, 73: 862-866)。HMBA由于诱导多种肿瘤细胞分化而抑制恶性变, 并选择性地改变癌细胞的基因表达, 进而通过鼠和犬的药代动力学和安全性实验, 经批准进入临床研究。Ⅱ期临床研究显示, 对骨髓增生异常综合征和急性髓性白血病有完全或部分缓解作用, 然而效果只是短暂的缓解。此外因HMBA的血浆半衰期t1/2为1.5 h, 需要不断地滴注给药, 以及血小板减少的不良反应, HMBA终止了研究(Breslow R, Jursic B, Yan ZF, et al. Potent cytodifferentiating agents related to hexamethylenebisacetamide. Proc Natl Acad Sci USA, 1991, 88: 5542-5546)。
3.4 辛二酸二酰胺化合物研究HMBA系列的同时, 还合成了它的“逆向酰胺”—二元酸酰胺化合物, 其中活性较高的化合物22和23, 分别为辛二酸与二甲胺和甲胺的酰胺, 诱导分化活性与HMBA相同, 在5 mmol·L-1浓度下诱导分化率 > 95%。对HL-60细胞的分化活性也与HMBA相当, 未进一步研究(Reuben RC, Khanna PL, Gazitt Y, et al. Inducers of erythroleukemic differentiation; relationship of structure to activity among planar-polar compounds. J Biol Chem, 1978, 253: 4214-4218)。
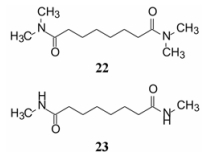
|
研究至此, 虽然HMBA等双酰胺类化合物的作用靶标仍不明确, 但构效关系已揭示出两个酰胺基团是优化的结构, 推测分别与两个相距不太远的位点结合, 通过螯合于金属离子或是发生氢键结合而起效, 但无论怎样的结合方式, 如果用异羟肟酸片段替换酰胺基有可能提高与金属的螯合力, 或增加氢键结合的机会, 因而研究了双异羟肟酸化合物。
4.1 柔性链连接的双异羟肟酸化合物参考HMBA的柔性链长, 合成了有代表性的二元酸的异羟肟酸化合物24~28和己二羟胺的双乙酰化物29。
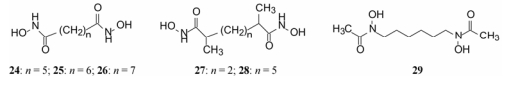
|
以苯环为连接基合成了刚性化合物p-苯二异羟肟酸(30)和m-苯二异羟肟酸(31), 半刚性分子p-苯二甲基异羟肟酸(32)和p-苯二乙基异羟肟酸(33), 还合成了含有不饱和键的化合物34和35, 考察刚性或刚柔并济化合物的基团空间取向对活性的影响。
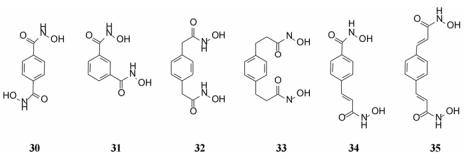
|
此外, 还设计合成了用五亚甲基或六亚甲基连接的双咪唑啉二酮化合物36和37, 这两个环状结构也包含有发生金属螯合或形成氢键能力的因素, 目的是考察成环对活性的影响。
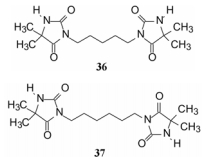
|
对上述含双异羟肟酸化合物评价了体外活性, 对小鼠红白血病细胞(MELC)的诱导分化作用列于表 2中(Richon VM, Webb Y, Merger R, et al. Second generation hybrid polar compounds are potent inducers of transformed cell differentiation. Proc Nat Acad Sci USA, 1996, 93 (3): 5705-5708)。
| Table 2 Activity of isohydroximic acids with flexible, semi-rigid, and rigid structures |
表 2列出的是有代表性的化合物, 旨在揭示有意义的构效关系, 概括如下: ①酰胺片段变换成异羟肟酸基团使诱导分化活性显著增强。化合物25是辛二酸双异羟肟酸, 活性高于18 (HMBA)和辛二酰双(二)甲胺(22和23) 100倍以上。②连接基换成庚二酸和壬二酸的双异羟肟酸(分别为24和26), 活性低于25; 缩短的连接基如27活性更弱, 提示结合的位点需要适宜位置的基团匹配, 这与上一节叙述的酰胺系列的构效关系相同, 也映射出双酰胺与双异羟肟酸的结合位点相同。③化合物28的酰基α位有甲基取代活性殆尽(与没有甲基取代的24比较), 提示可能为位阻效应所致, 这在双酰胺系列中未出现这个现象。④刚性的p-苯二异羟肟酸(30)的活性与25的相当, 但单个异羟肟酸则无活性(表 2未列出), 将第二个异羟肟酸连接到间位, m-苯二异羟肟酸(31)仍无活性, 再引入一个成间苯三异羟肟酸也无活性(未列出)。提示了两个异羟肟酸的空间位置对于结合位点的匹配非常重要。⑤化合物30的两边各嵌入一个亚甲基, 32失去活性; 各嵌入两个亚甲基则恢复活性; 含有双键的化合物35活性相当强, 即使只在单边引长取代基的化合物34, 虽然分子长度与无活性的32相同, 但活性仍很强, 微小的结构变动引起活性的剧烈变化, 提示结合的精准与复杂性。⑥这类化合物结合的复杂性还表现在, 虽然柔性化合物35与刚性的30的活性都很强, 但结合的位点是不同的, 当用低于有效浓度的这两个化合物共同温孵细胞时, 并未产生有效的诱导作用, 说明二者没有加和作用, 推论结合位点是不同的(两个柔性的化合物之间有加合作用, 数据省略)。
对上述活性较强的化合物进一步评价对HL-60和HT-29细胞的诱导分化作用, 表明化合物25 (简称SBHA)活性最好。SBHA与化合物HMBA (18)作结构比较, 两端功能基之间都是由6个亚甲基相连, 分子长度相近; 酰胺基与异羟肟酸基都是金属螯合基团, 也都是氢键的给体和接受体。诱导分化性质也有加和性, 似乎作用机制相同。事实上, 当发现HDAC靶标后的研究表明, 二者的作用靶标是不相同的。但这是后话。
5 杂合的双极性化合物SBHA和HMBA分别为含有双异羟肟酸基和酰胺基化合物, SBHA的活性更强。由于羟肟酸是金属强螯合基团, 但分子中存在两个强力螯合基团未必是必需的。因而保留一个, 另一用其他极性基团置换, 即杂合的双极性分子, 共合成了600多个化合物(Breslow R, Marks PA, Rifkind RA, et al. Novel potent inducers of terminal differentiation and methods of use thereof. PCT Int Appl. WO 93107148, 1993-04-15)。结构变换中的一个重要考虑是, 在非羟肟酸一端的极性基团上, 连接疏水性基团, 可利用酶或受体活性部位的疏水腔或裂隙, 发生疏水-疏水相互作用, 以增强活性, 例如化合物1和38。1是辛二酰苯胺异羟肟酸, 简称SAHA, 38简称Cl-UCHA (Meinke PT, Liberator P. Histone deacetylase: a target for antiproliferative and antiprotozoal agents. Curr Med Chem.2001, 8:211–235; Richon VM, Emiliani S, Verdin E, et al. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Natl Acad Sci USA, 1998, 95: 3003-3007)。
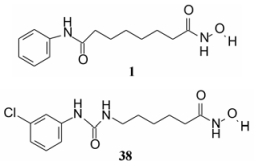
|
组蛋白去乙酰化酶(HDAC)的发现和结构解析对理解上述的研发轨迹豁然开朗。HDAC是细胞核内催化乙酰化的组蛋白脱乙酰基, 降低组蛋白乙酰化程度的酶系。与HDAC相对立的另一酶系是组蛋白乙酰化酶(HAT), 二者功能完全相反, HAT是促进组蛋白乙酰化的酶系, 将乙酰辅酶A的乙酰基转移到组蛋白氨基末端特定的赖氨酸残基的ε-氨基上, 由于氨基被乙酰化, 失去形成正电荷的能力。HDAC与之相反, 它使组蛋白去乙酰化, 暴露出氨基, 并形成正电荷, 与带负电荷的DNA紧密结合, 使染色质致密卷曲, 导致基因的转录受到抑制。正常细胞的HAT和HDAC处于动态平衡。在癌细胞中, HDAC过度表达导致去乙酰化作用的增强, 增加了DNA与组蛋白之间的结合力, 使松弛的核小体变得十分紧密, 不利于特定基因的表达, 包括一些肿瘤抑制基因的表达。抑制HDAC可提高染色质特定区域组蛋白乙酰化, 调控细胞凋亡及分化相关蛋白的表达和稳定性, 诱导细胞凋亡及分化, 因而HDAC是肿瘤治疗的一个靶标。
6.2 天然产物曲古抑菌素A的抑制HDAC作用天然产物曲古抑菌素A (2, trichostatinA, TSA)是链霉菌的代谢产物, 起初作为抗真菌剂, 后来发现可抑制真核细胞周期初期的生长阶段, 促进与凋亡相关的基因表达, 抑制癌细胞生长。2对组蛋白去乙酰化酶(HDAC)呈强效抑制, 无细胞抑制HDAC的IC50为1.5 nmol·L-1 (Finnin MS, Donigian JR, Cohen A, et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature, 1999, 401: 188-193; Yoshida M, Kijima M, Akita M, et al. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem, 1999, 265: 17174-17179)。
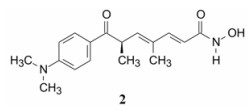
|
鉴于SAHA与TSA的结构相似性, 特别是分子的形状、尺寸和药效团特征的分布也相似, 因而评价了SAHA以及前述的双极性分子对HDAC的活性。结果表明SAHA (1)对HDAC1和HDAC3的抑制活性与TSA相当, IC50达到1~5 nmol·L-1。SBHA (25)对HDAC也具有强抑制活性。然而HMBA (18)却对HDAC没有显示活性, 提示双酰胺化合物的分子形状虽然与双酰胺化合物SAHA相似, 也具有诱导细胞分化作用, 但作用靶标不是HDAC。由此可见, 化合物之间具有相同的细胞或生理表型变化未必意味着作用靶标是相同的(Richon VM, Emiliani S, Verdin E, et al. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Nat Acad Sci USA, 1996, 95: 3003-3007)。
7.2 SAHA与酶活性部位的结合模式SAHA与HDAC样蛋白(相当于HDAC的催化部位)的复合物晶体结构表明(图 2), 酶的催化部位是窄长形的结合腔, SAHA通过亚烷基链将异羟肟酸基团引向腔体的深部, 与该处的锌离子形成双齿型配位键结合, 分子另一端的苯基则处于酶的疏水面, 发生疏水-疏水相互作用。天然产物TSA (2)与HDAC的结合模式与SAHA相同(Finnin MS, Donigian JR, Cohen A, et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature, 1991, 401: 188-193)。

|
Figure 2 Crystallographic diagram of the complex of SAHA with HDAC active site |
体外实验表明SAHA对多种肿瘤细胞(包括耐药株)有抗增殖和促凋亡作用, 并抑制血管生成。与其他抗肿瘤药物合并应用, 可提高动物移植性肿瘤的敏感性, 呈现显著的协同作用(Campbell R, Sanchez E, Steinberg J, et al. The potent histone deacetylase inhibitor vorinostat, in combination with melphalan, markedly enhances the anti-myeloma effects of chemotherapy in vitro and in vivo. Cancer Res, 2008, 68: 733)。药代动力学实验表明SAHA可口服吸收, 半衰期t1/2 2 h, 与血浆蛋白结合率71%。SAHA不与CYP发生相互作用。体内代谢途径主要是葡醛酸苷化, 也发生水解和脂肪酸的β-氧化。代谢产物没有活性, 主要从尿中排泄。基于临床前SAHA的安全有效性实验, 确定为候选化合物, 定名为伏立诺他(volinostat), 经临床研究, 表明是治疗复发性皮肤T性病淋巴瘤的有效药物, 于2007年经FDA批准在美国上市(Marks PA, Breslow RB. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol, 2007, 25: 84-90)。
8 后继的HDAC抑制剂 8.1 首创药物难, 跟随式药物也不易伏立诺他是在靶标或作用机制没有任何借鉴的情况下, 由简单的DMSO分子演化成后来揭晓的以HDAC为靶标的首创性抗肿瘤药物, 这是完全按照药物化学和构效关系的方法, “摸着石头到达了河的彼岸”, 完成了新药的研发, 实现了由表型评价到靶标的确认。后继的抑制剂研究由于有了TSA、伏立诺他以及HDAC晶体结构的多重依据, 跟随式的研究应比伏立诺他的历程容易的多。然而难度仍然很大, 开发跟随性的新药需要超越首创的药物, 以获得临床和市场的认可, 风险的置后使难度加大。就HDAC靶标而言, 对酶亚型的特异性和对组织器官的选择性是超越前驱的关键所在。因此自伏立诺他上市10年来尚无第二个HDAC抑制剂的药物问世。
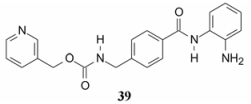
8.2 恩替诺特恩替诺特(39, entinostat)是目前最接近成功的候选药物。其化学结构的特点是用邻苯二胺的酰胺作为异羟肟酸的生物电子等排体, 保持与锌离子的螯合作用, 对HDAC1和HDAC3的抑制活性IC50分别为0.51和1.7 μmol·L-1。恩替诺特用于治疗乳腺癌, 现处于临床Ⅲ期研究阶段(Suzuki T, Ando T, Tsuchiya K, et al. Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives. J Med Chem, 1999, 42: 3001-3003; Heltweg B, Dequiedt F, Marshall BL, et al. Subtype selective substrates for histone deacetylases. J Med Chem, 2004, 47: 5235-5243)。
|
由于伏立诺他等HDAC抑制剂的构效关系清晰明确, 加之抑制剂与HDAC复合物的晶体结构的解析, 设计合成了多种结构类型的活性化合物。尤启冬等综合多种HDAC抑制剂, 以TSA (2)与HDAC复合物晶体结构中的构象为基础, 优化其他抑制剂的活性构象, 构建了药效团模型如图 3所示。图中药效团的模型分子是伏立诺他, 红色骨架是伏立诺他在复合物晶体结构的构象, 绿色骨架是生成药效团的伏立诺他构象, 二者相似性很高。其中重要的因素是在端点为两个氢键接受体, 即锌结合基(ZBG), 与酶结合部位的辅因子锌离子形成双齿型配位键。三个疏水结合位点的端点帽区(疏水位点3)与酶开口处的疏水区发生相互作用(Chen YD, Li HF, Tang WQ, et al. 3D-QSAR studies of HDACs inhibitors using pharmacophore-based alignment. Eur J Med Chem, 2009, 44: 2868-2876)。

|
Figure 3 The pharmacophore of HDAC inhibitors |