 2018, Vol. 53
2018, Vol. 53



肿瘤免疫治疗是一种通过恢复机体正常的抗肿瘤免疫反应控制和清除肿瘤的治疗方法。吲哚胺2, 3-双加氧酶1 (IDO1)是一个含亚铁血红素的酶, 是色氨酸代谢的关键酶, 在多种肿瘤细胞中过度表达, 消耗肿瘤微环境中的色氨酸, 造成T细胞抑制, 从而使肿瘤细胞逃避免疫系统的监控和清除, 是肿瘤免疫治疗的一个重要靶点[1]。
色氨酸是人体必需氨基酸, 有两条代谢途径:犬尿氨酸代谢途径和褪黑素途径。其中犬尿氨酸途径占95%, 是主要的代谢途径, 会产生多种生物活性小分子。犬尿氨酸代谢途径的第一步也是限速步骤, 由IDO1、IDO2和色氨酸2, 3-双加氧酶(TDO)三种酶催化。IDO1是一种肝外细胞溶质酶, 分布在人体各个组织中, 包括胎盘、脑、肺、肝、脾、肾、胃、大肠、小肠、结肠等[2], 发挥主要的催化作用[1, 2]; IDO2主要分布在肾小管、肝脏和精子中[1, 3], 色氨酸代谢活性低; TDO主要在肝中表达, 与IDO催化的反应相同, 调控血液中色氨酸的浓度[1]。
1 IDO1的结构2006年, IDO1复合物的晶体结构(2D0T、2D0U)首次被报道[4], 揭示了IDO1蛋白结合腔的特征, 为理性分子设计提供了依据。IDO1是一种含亚铁血红素的双加氧酶, 包括两个结构域。其中大的结构域是由13个α-螺旋(G-S)和两个310螺旋组成的全螺旋结构域。4个长螺旋(G、I、Q、S)与血红素平面大致平行, 并与其他3个短螺旋(K、L、N)共同构成了血红素结合口袋(图 1A)。螺旋Q中的His346的咪唑是血红素中铁离子的内源性配体, 在第五配位位置与铁离子络合(图 1B)。小的结构域在血红素的远侧, 由6个α-螺旋(A~F)、两个短β-折叠和3个310螺旋构成, 位于血红素铁离子第六配位位置的上方, 覆盖血红素口袋的顶部(图 1A)。

|
Figure 1 The crystal structure of IDO1-4PI complex (PDB code: 2D0T) |
由IDO1复合物的晶体结构(图 1B)可以看出, IDO1结合腔包括A、B两个结合口袋, 其中A腔位于铁离子第六配位位置上方, 是由Ala264、Gly262、Tyr126、Val130、Cys129、Phe163和Phe164等氨基酸构成的疏水性口袋, B腔位于开口处, 由Phe226、Phe227、Arg231、Ile354和Leu384等氨基酸构成。氨基酸突变实验表明, His346、Asp274、Phe226、Phe227、Arg231是IDO1蛋白的关键氨基酸, 与蛋白的稳定性、催化活性和底物识别有关[4, 5]。研究发现当将Asp274突变成Ala时, 会影响IDO1的催化活性[5], 这是由于Asp274与Arg343形成盐桥, 而Arg343位于血红素卟啉环附近与6位丙酸基形成氢键从而稳定血红素。Phe226、Phe227、Arg231通常与配体形成π-π或者阳离子-π相互作用, 从而对IDO1的催化活性和底物识别产生影响。Ser167Ala的突变实验表明, Ser167既不位于底物识别区, 也不会影响酶的催化活性[4], 然而许多晶体结构都证实, Ser167会与配体形成氢键作用, 是IDO1结合口袋的关键氨基酸[6, 7]。
2 IDO1的生物学功能IDO1是色氨酸沿犬尿氨酸途径代谢的限速酶, 是代谢途径的第一步[8]。如图 2所示, 在IDO1的催化下, 色氨酸中吡咯环发生氧化裂解, 生成N-甲酰犬尿氨酸, 经犬尿氨酸甲酰胺酶去甲酰化生成犬尿氨酸, 在犬尿氨酸酶的催化裂解下生成邻氨基苯甲酸, 随即转化为2-氨基黏康酸、2-吡啶甲酸和喹啉酸, 喹啉酸在磷酸核糖转移酶的作用下生成NAD+。IDO1通过催化色氨酸的氧化裂解反应, 调控体内色氨酸及代谢产物的水平, 发挥着重要的生物学功能。

|
Figure 2 The kynurenine metabolic pathway |
IDO1介导的肿瘤免疫逃逸是综合调节多种免疫细胞的结果, 其中最重要的是T细胞[9, 10]。包含两方面的作用, 一是抑制效应T细胞、NK细胞等免疫杀伤性细胞活性或诱导其凋亡[11, 12]; 二是促进致耐受性DC细胞的分化, 促进Treg细胞、髓系衍生抑制细胞(MDSC)等免疫负向调节细胞的增殖和提高其活性[13], 二者协同作用产生免疫抑制, 帮助肿瘤细胞逃避免疫系统的杀伤。
如图 3所示, IDO1发挥免疫抑制作用的分子机制主要是通过调节色氨酸的代谢, 导致局部色氨酸含量降低和犬尿氨酸及其他代谢产物含量升高。在肿瘤微环境中, 肿瘤细胞自身可分泌IDO1, 同时会招募周围的基质细胞及其他免疫细胞(APC、MDSC、CAF、巨噬细胞等)表达IDO1, 过表达的IDO1会使肿瘤局部色氨酸水平降低, 犬尿氨酸及其他代谢产物水平升高。色氨酸的缺乏会通过多种分子机制抑制T细胞的活性, 包括mTORC1途径和GCN/eIF2途径等[14-16]。色氨酸含量下降会抑制T细胞中的mTORC1, 抑制T细胞活性, 诱导T细胞发生自噬; 另外一个分子机制是色氨酸的缺乏导致不带电荷的色氨酸转运核糖核酸(tRNA)在细胞中积累, 激活GCN2, 激活的GCN2使eIF2α磷酸化, 进而抑制eIF2β, 抑制蛋白的合成, 阻止T细胞的活化。此外, 激活的GCN2也能促进Treg细胞的分化并增强活性[15]。同时, 色氨酸代谢导致犬尿氨酸及其他代谢产物增多, 犬尿氨酸作为AHR内源性的配体, 可以活化AHR的转录, 从而促进免疫抑制性白介素-10 (IL-10)的表达, 促进FOXP3+Treg细胞的分化, 最终导致T细胞沉默, Treg细胞被激活[17-19]。上述多个信号通路的共同作用, 导致肿瘤微环境产生免疫抑制作用, 促进了肿瘤细胞的免疫逃逸。

|
Figure 3 Mechanism of IDO1 regulating T cell function and activity |
近年来, IDO1抑制剂的研究得到了广泛性的关注, 吸引了众多制药公司和科研高校参与, 采用高通量筛选方法及基于结构的药物分子设计手段, 发现了多种结构类型的IDO1抑制剂。目前已有10个IDO1小分子抑制剂进入临床研究阶段(表 1), 但仍没有药物上市。Incyte公司开发的Epacadostat和百时美施贵宝公司开发的BMS986205进入临床Ⅲ期研究阶段, 另外NewLink公司研发的Indoximod目前处于临床Ⅱ/Ⅲ期研究阶段。已有研究者对IDO1的生物学功能及抑制剂的研究进行了综述[20-25], 本文将根据化合物的结构类型, 对代表性IDO1小分子抑制剂的最新研究进展进行综述。
| Table 1 IDO1 inhibitors in clinical trials |
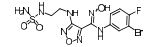
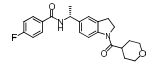
如图 4所示, 2009年, Incyte公司通过高通量筛选获得了IDO1选择性竞争性抑制剂1 (Ki = 1.5 μmol·L-1)。光谱学实验证实了苗头化合物1可以与血红素中的铁离子直接络合。对化合物1进行结构改造, 发现取代的苯胺替换苄胺, 可显著提高化合物的抑制活性, 获得了高活性的IDO1抑制剂2 (INCB14943)。研究者对化合物2进行了体内活性和药代动力学性质评价, 发现小鼠每天给药两次, 每次75 mg·kg-1时, 抑瘤率最高可达50%[26], 但是化合物2口服半衰期短(t1/2 < 0.5 h)。为了解决化合物2口服生物利用度低、半衰期短的缺陷, 研发者对化合物2继续进行结构改造, 期望获得药代性质优良的类药化合物。结构改造集中在伸向B口袋的氨基, 发现氨基上双取代(NMe2)时, 对IDO1抑制活性消失; 氨基上单取代(NHMe、NHEt、NHBn)时, 活性保留或稍有降低, 清除率降低, 但蛋白结合率高。为了降低化合物的血浆蛋白结合率, 研究人员在氨基上引入极性侧链, 获得了理化性质较好的候选药物3 (epacadosat)。采用CT26移植瘤模型小鼠, 每天口服给药30 mg·kg-1时, 给药13天, 抑瘤率可达56%。毒理和药代实验表明, 候选药物3安全性高、药代性质良好(犬和灵猴的口服生物利用度分别为59%和33%)[27]。2015年11月, epacadosat用于治疗黑色素瘤进入临床Ⅰ期研究阶段。遗憾的是, 2018年4月公布的Ⅲ期临床ECHO-301的数据显示, epacadostat联合PD-1单克隆抗体pembrolizumab治疗不可手术切除或者转移性黑色素瘤, 相比单独使用pembrolizumab, 无进展生存时间(PFS)无明显改善。目前, epacadostat联合pembrolizumab一线治疗非小细胞肺癌适应证开发, 进入Ⅱ期临床研究阶段。Incyte公司将继续开展epacadostat联合疗法(除PD-1抗体外)在多个肿瘤的早期临床研究, 这里的联合疗法主要包括epacadostat + nivolumab + ipilimumab、epacadostat联合疫苗、epacadostat联合化疗疗法等。

|
Figure 4 The optimization process of epacadostat (compound 3) |
2017年, Syun-Ru Yeh课题组[7]获得了epacadostat与IDO1复合物的晶体结构(PDB code: 5WN8)。如图 5所示, 从该晶体结构中可以清楚地看到, epacadostat中羟基脒的氧原子与血红素中心的铁离子形成配位键, 同时与Ala264的NH形成氢键相互作用。卤素取代的芳环伸向了A腔, 与周围氨基酸形成疏水相互作用, 芳环上的氟原子(4-F)与Cys129上的巯基形成了较强的氟-硫相互作用。噁二唑环4-位氨基上引入的极性侧链伸向B腔, 磺酰基上的氧原子与Arg231形成氢键。此外, 羟基脒上的氮原子与噁二唑上的氨基、羟基脒上的氧原子及芳胺上的NH分别形成了分子内氢键, 不仅使得该类化合物具有良好的膜渗透性和口服生物利用度, 同时使得化合物具有一定的刚性, 降低了化合物与IDO1结合时的熵损失, 有利于提高分子的活性。

|
Figure 5 The binding mode of epacadostat with IDO1 (PDB code: 5WN8) |
如图 6所示, 基于epacadostat的结构, 研究者在伸入B腔的极性侧链上进行结构改造, 获得了结构新颖、活性相当或者稍有提高的IDO1抑制剂(4[28]、5[29])。维持羟基脒结构不变, 改变中间连接基团, 将噁二唑环替换成哌啶环(6)[30]、苯并咪唑环(7)[31]、苯并噁二唑(8、9)[32], 或者将噁二唑替换成疏水性脂肪环(10)[33], 均获得了具有较好抑制活性的IDO1抑制剂。

|
Figure 6 Some examples of N-hydroxyamidine-based IDO1 inhibitors |
该类化合物构效关系研究表明: ①占据A腔的苯环上3-位引入卤素活性较好, 3, 4-位双卤素取代时, 酶学水平和细胞水平抑制活性可以保持, 且体内清除率显著降低[26, 27]; ②当羟基脒的N-OH替换成N-OMe、NH、N-NH2、O、S后, 活性均消失, 提示羟基脒与铁离子的络合对活性贡献显著[26]; ③中间连接片段噁二唑环可以替换; ④伸向B腔的磺酰脲侧链可以替换成其他极性基团, 侧链的长度也有较高的容忍度, 中间碳原子可以维持在1~4个[28, 29], 可以利用侧链调整化合物的理化性质和药代动力学性质。
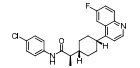
4.2 喹啉类IDO1小分子抑制剂2015年BMS (百时美施贵宝)收购Flexus Biosciences, 获得了其IDO1小分子抑制剂的开发权力, 即BMS-986205 (图 7, 化合物11)。BMS-986205[34]是一个可口服的IDO1小分子抑制剂, 与PD-1抑制剂nivolumab联合用药, 处于临床Ⅲ期研究阶段。如图 7所示, 将化合物11中的酰胺键反转(12)或者替换成脲基(13)、磺酰胺基(14)[35]时, 所得化合物均有较强的抑制活性; 当喹啉环替换成三氟甲基取代的吡啶环(16)时[27], 抑制活性仍然保留; 当以4-羟基哌啶作为连接基团时, 活性降低(15、17)[36]。值得关注的是, 与其他类型IDO1抑制剂不同, 该类化合物是与不含亚铁血红素的IDO1蛋白结合, 具有独特的结合模式, 如图 8所示: 4-氰基取代的苯环位于A腔, 与Tyr126形成π-π相互作用, 酰胺上的NH与Ser167的侧链OH形成氢键作用; 环己烷作为连接基团, 将喹啉环导向一个新的疏水性结合腔, 喹啉环占据了由Phe270、Phe214、His346及Arg343等组成的结合口袋, 与Phe270形成π-π相互作用, 喹啉上的氮原子与Arg343的侧链NH形成氢键作用[37]。该类化合物独特的结合模式, 是否会产生独特的药效学特征, 还有待于临床结果验证。这种结合模式, 为设计结构多样的IDO1抑制剂提供了新的思路。

|
Figure 7 Representative structures of quinolones |

|
Figure 8 The binding mode of BMS-116 with IDO1 (PDB code: 6AZW) |
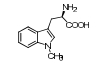
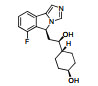
PF-06840003是由iTeos公司发现, 辉瑞公司联合开发的一个高选择性的可口服的IDO1抑制剂。如图 9所示, PF-06840003的苗头化合物19是通过高通量筛选发现的, 对hIDO1具有中等抑制活性(IC50 = 3.0 μmol·L-1), 且对TDO无抑制活性, 是一个高选择性的hIDO1抑制剂。以化合物19作为苗头化合物, 分别对吲哚3-位的琥珀酰亚胺、吲哚骨架、4, 7-位取代基、5, 6-位取代基进行结构优化。研究结果表明, 琥珀酰亚胺对抑制活性贡献显著; 当吲哚环的1, 2, 4, 7位分别被不同基团取代时, 活性均显著降低或消失; 当采用其他环系替换吲哚骨架时, 仅萘环替换吲哚环时, 所得化合物具有抑制活性, 但活性有所降低; 在吲哚的5-位引入卤素时活性明显提高, 且F > Br > Cl (5-F、5-Br和5-Cl的IC50分别为0.15、0.37和0.83 μmol·L-1); 6-位引入不同的基团时, 仅氰基(IC50 = 1.7 μmol·L-1)和Br (IC50 = 0.42 μmol·L-1)取代时活性有所提高; 但当5-氟-6-溴双取代时, 活性较5-氟单取代略有降低。因此, 选择了5-氟取代的化合物20作为候选药物。酶学活性评价表明, R构型化合物20a (IC50 = 0.12 μmol·L-1)是一个高选择性的hIDO1抑制剂, 而S构型的20b (IC50 = 54 μmol·L-1)对hIDO1抑制活性弱。然而在细胞水平活性评价中, 20b (HeLa cell, IC50 = 13 μmol·L-1)仅比20a (HeLa cell, IC50 = 1.0 μmol·L-1)的活性低13倍, 推测可能是由于手性碳原子可以通过烯醇式互变, 导致20b部分消旋, 活性提高。因此, 选择了消旋体20进入临床前和临床研究[6]。

|
Figure 9 The optimization process of PF-0684003 |
化合物PF-06840003与IDO1的结合模式如图 10所示, 吲哚环占据了IDO1的疏水性口袋A腔, 1位的NH与Ser167形成氢键作用, 苯环与Tyr126、Phe163、Phe164形成π-π相互作用; 3位琥珀酰亚胺上的NH与卟啉上7-丙酸基形成氢键, 两个羰基分别与Ala264、Thr379形成氢键; 这些氢键网络的形成及吲哚环与结合口袋之间的疏水相互作用, 使得该候选药物虽然没有与铁离子络合, 但仍具有较高的抑制活性[6]。

|
Figure 10 The binding mode of PF-06840003 with IDO1 (PDB code: 5WHR) |
PF-06840003是一类不与铁离子络合的hIDO1小分子抑制剂, 具有适宜的酶学水平抑制活性(IC50 = 0.15 μmol·L-1)、高配体效率(LE = 0.53)、低血浆蛋白结合率(fu = 0.45)、良好的药代性质, 可预测的半衰期为19 h, 生物利用度为64%, 目前正在美国开展临床Ⅰ期试验, 用于治疗神经胶质瘤。
4.3.2 Indoximod和其他吲哚类化合物1991年报道的DL-1MT (图 11, 化合物21)[38, 39]是弱的IDO1抑制剂, 手性拆分后, 发现L-1MT对IDO1的酶抑制浓度Ki值为34 μmol·L-1[39], 而D-1MT对IDO1无抑制作用, 但体内实验结果表明, 与L-1MT相比, D-1MT (indoximod)单用或者与化疗药物合用时, 具有更好的抗肿瘤活性。目前普遍认为indoximod的抗肿瘤机制是其可以调节色氨酸跨膜运输[40], 另外模拟色氨酸调控IDO1下游信号通路, 被称为IDO1旁路抑制剂[41]。Indoximod作为色氨酸的类似物, 使T细胞中因色氨酸减少而被抑制的mTORC1脱阻抑, 从而上调效应T细胞中的共调节受体ICOS, 发挥免疫调节作用[42-44]。Indoximod由NewLink Genetic公司开发, 目前正在进行临床Ⅱ/Ⅲ期研究, 主要与其他肿瘤免疫治疗药物pembrolizumab或者nivolumab联用, 用于治疗转移性前列腺癌、急性髓样白血病、原发性恶性脑肿瘤、转移性胰腺癌、转移性乳腺癌、转移性黑色素瘤、非小细胞性肺癌等。

|
Figure 11 Some examples of indole analogs |
2017年, Syun-Ru Yeh课题组[7]通过引入氰基与铁离子络合, 获得了色氨酸与IDO1复合物的晶体结构, 如图 12所示, 氰基上的氮原子分别与Ala264、吲哚NH及色氨酸上的氨基形成氢键相互作用。色氨酸的吲哚环占据了A腔, 吲哚NH通过一分子水介导与Ser167形成氢键作用。吲哚3-位侧链伸向B腔, 侧链上的氨基与卟啉环的7-丙酸基及Thr379形成氢键网络; 羧基通过与Arg231及Thr379形成离子键进一步稳固结合。

|
Figure 12 The crystal structure of IDO1-Trp complex (PDB code: 5WMU) |
众所周知, IDO1内源性配体为色氨酸, 所以早期IDO1抑制剂多为吲哚或色氨酸衍生物, 如图 11所示。这些化合物多数采用普筛方法, 获得苗头化合物, 通过结构改造, 获得抑制活性在微摩尔水平的IDO1抑制剂(22~26)[45-50]。可能是由于多数吲哚类化合物缺少与铁离子络合的基团, 因此对IDO1的抑制活性均不高。此外, 将吲哚骨架用生物电子等排体吲唑(27)[51, 52]或吡啶并咪唑(28)[53]替换后, 4-位引入疏水性基团伸向B腔, 活性有所提高, 特别是化合物28, 对IDO1的抑制活性达到纳摩尔水平。
4.4 含咪唑或三氮唑类化合物 4.4.1 Navoximod (NLG919)1989年, 4-苯基咪唑(29, 4-PI, IC50 = 48 μmol·L-1)被确认为是弱的非竞争性IDO1抑制剂。如图 1B所示, 2006年报道了4-PI与IDO1复合物的晶体结构(2D0T)[4], 使得该结构类型IDO1小分子抑制剂的研究再一次成为热点。晶体结构显示, 4-PI占据了IDO1疏水性结合口袋A腔, 咪唑1位NH位于铁离子第六配位附近, 与铁离子络合, 苯环与Phe163形成π-π相互作用。在该复合物晶体结构中, 两个反向平行的CHES (2-环己胺基乙磺酸)占据了B腔, 与卟啉环上的7-丙酸基形成氢键。该结构信息提示, 可以在4-PI结构的基础上, 引出侧链, 伸向B腔, 利用其与卟啉环上7-丙酸基的氢键作用及B腔的疏水作用提高抑制剂与IDO1的结合强度。随后, 基于复合物的晶体结构(2D0T), 多个团队[54-56]对4-PI进行了结构改造, 可以归结为三方面: ①在咪唑的N-1、C-2、N-3位引入基团, 占据CHES所在的空腔, 提高活性; ②在苯环的邻、间、对位分别引入小体积的取代基, 与A腔中的Cys129、Ser167产生相互作用, 增强结合; ③利用生物电子等排策略, 优化铁离子络合基团。如图 13所示, 该类抑制剂构效关系结果显示: ① N-3位可以引入苄基(30, IC50 = 32 μmol·L-1); ②苯环邻位引入羟基, 可与Ser167形成氢键, 显著提高活性(31, IC50 = 4.8 μmol·L-1); ③咪唑、三氮唑(32, IC50 = 86 μmol·L-1)[56]可以与铁离子络合。在化合物32的基础上, 对三氮唑类化合物的苯环上取代基进行结构优化, 获得IDO1抑制剂33, 抑制活性在1×10-7 mol·L-1水平, IC50值为0.33 μmol·L-1[57]。

|
Figure 13 The optimization process of navoximod |
在咪唑类IDO1小分子抑制剂的结构改造中, NewLink公司做了很多尝试。在确定咪唑N-3和苯环2'位可以引入基团后[55], 在2'位通过氧原子引入脂肪性或芳香性含杂原子的疏水性侧链(34, IC50 < 1 μmol·L-1), 通过增强在B腔的结合来提高活性; 同时尝试将N-3位与2'位通过稠环连接, 合成三环类化合物35, IC50值在1~10 μmol·L-1, 活性得到保留[58]。在三环骨架上, 引入侧链, 伸向B腔[59], 结构优化后, 获得候选药物navoximod (36, IC50 = 13 nmol·L-1)。Navoximod小鼠口服给药时, 可使血浆和组织中的犬尿氨酸浓度下降50%, 同时可以促进效应T细胞的增殖, 使肿瘤体积减少。在重复给药毒性实验中, 连续给药28天, 每天给药两次, 剂量为80 mg·kg-1, 无明显毒副作用[60]。2014年, 罗氏制药公司旗下的Genetech与NewLink公司签署协议合作开发navoximod, 遗憾的是, navoximod联合紫杉烷类化疗药(多西紫杉醇或紫杉醇)治疗转移性乳腺癌Ⅱ期临床研究结果并不理想。数据显示, 与紫杉烷单用相比, navoximod联合紫杉烷化疗在无进展生存期、总生存期、客观缓解率方面均未表现出统计学显著性差异。2016年Wu Su-Ying及其团队[61]获得了NLG919类似物与IDO1复合物的晶体结构(图 14, PDB code: 5EK3)。从该结构中可以看出, 异吲哚并咪唑环占据了结合口袋的A腔, 咪唑的N原子与铁离子络合, 环己烷伸向B腔, 与周围氨基酸形成疏水作用。侧链上的羟基与异吲哚上的N原子形成分子内氢键, 同时与7-丙酸基形成氢键网络。分子内氢键的形成, 对于稳定构象、提高活性及改善化合物的理化性质发挥着重要的作用。当用羰基代替羟基或者将五元环替换成六元环, 不能形成分子内氢键, 化合物的抑制活性显著降低甚至消失。该化合物含有两个手性中心, 为4个立体异构体的混合物。手性拆分后, 分别测试异构体的活性, 发现异吲哚环上C-1位为S构型的两个异构体具有较强的的抑制活性, 这与晶体结构的解析结果一致。

|
Figure 14 The binding mode of NLG919 analogue with IDO1 (PDB code: 5EK3) |
随着navoximod进入Ⅰ期临床研究, 该类化合物的衍生物也相继出现。对伸向B腔的疏水性基团及三环骨架进行结构改造, 获得了新结构的高活性IDO1小分子抑制剂(图 15, 37~45)[62-67]。其中37~42是维持三环骨架不变, 改造深入B腔的环己基及中间连接基团, 伸向B腔的大体积的疏水性基团可以是脂肪环或者芳环, 允许较大的结构变换, 有利于提高化合物的结构新颖性和药代性质。化合物43~45则主要对三环骨架进行改造, 苯环替换成吡啶[65]、噻吩[66]或者改变咪唑环上N原子的位置[64]均得到了较强抑制活性的IDO1抑制剂。

|
Figure 15 Some examples of imidazole and triazole IDO1 inhibitors |
4-氨基三氮唑化合物46 (图 15)是一个新结构骨架的IDO1抑制剂, 在细胞水平具有显著的抑制活性。如图 16所示, 其与IDO1复合物的晶体结构(PDB code: 6F0A)显示, 化合物46的结合模式与4-PI类似, 主要占据A腔, 对氯苯基与周围氨基酸Tyr126、Cys129、Val130、Gly262、Phe164等形成疏水相互作用, 三氮唑1-位N与卟啉环上铁离子络合, 4-位氨基与Ser167形成氢键作用。化合物46细胞活性较酶学活性高出近500倍, 主要是由于化合物46与IDO1的Fe2+会缓慢的形成稳固的结合, 解离速度慢。相较于酶学水平实验, 细胞水平实验具有适宜的pH、较长的孵育时间及还原性的环境, 使化合物46与IDO1的Fe2+形成牢固的结合[68]。

|
Figure 16 The binding mode of 4-aminotriazole 46 with IDO1 (PDB code: 6F0A) |
2014年日本住友制药有限公司(Dainippon Sumitomo Pharma Co, Ltd.)报道了化合物47 (图 15, Amg-1)与IDO1复合物的晶体结构(图 17A, PDB code: 4PK5)[69]。其噻唑并三氮唑环上的氮原子与铁离子络合, 对甲基苯基占据了A腔, 酰胺键延伸出的侧链亚甲二氧基苯基占据了B腔, 与关键氨基酸Phe226和Arg231形成相互作用。由于配体的碱性对形成金属络合物至关重要, 当采用咪唑代替三氮唑, 提高化合物的碱性, 侧链仍采用4个原子长度的刚性连接基团, 优化得到化合物48 (图 15, IC50 = 77 nmol·L-1), 活性提高近40倍, 伸向B腔的苯环与Phe226形成π-π相互作用, 4位的氰基与Arg231形成静电相互作用。如图 17B所示, 与化合物48和IDO1复合物的晶体结构(PDB code: 4PK6)相比, 化合物47以完全不同于化合物48的延展构象结合于B腔, IDO1的结合口袋呈现了明显的诱导契合, 主要表现在Ala260-Ser263链的移动及Phe226和Arg231侧链的摆动, 导致结合口袋B腔形状和大小明显改变。

|
Figure 17 (A) The binding mode of Amg-1 with IDO1 (PDB code: 4PK5); (B) Superposition of 4PK5 (gold) and 4PK6 (blue) |
除上述进入临床研究阶段的IDO1抑制剂外, 还有其他结构类型多样的IDO1抑制剂被报道(图 18)。其中苯磺酰肼化合物49是通过对苯基咪唑4-PI的结构优化得到的, 是一个活性、选择性和口服生物利用度等性质均较优的IDO1抑制剂[70, 71]。化合物50是复旦大学通过高通量筛选获得的活性较好的1-茚酮类抑制剂, 结构改造发现去除或改变酮羰基的位置会使抑制活性降低, 推测1-茚酮类化合物的酮羰基可能与IDO1的血红素中心Fe2+配合[72]。色胺酮衍生物51在酶和细胞IDO1抑制试验、T细胞增殖试验、表面等离子体共振结合试验、Lewis肺癌肿瘤小鼠试验中均显示活性[73]。澳大利亚悉尼新南威尔士大学的Andrew课题组推测消炎镇痛药依布硒啉52通过与IDO1蛋白上的多个半胱氨酸残基形成硒硫共价键结合, 降低IDO1蛋白的螺旋含量和稳定性, 进而抑制IDO1酶活性[74]。氨基腈类化合物53[75]和二氨基取代呋喃吡啶化合物54[76]均由Curadev制药公司研发, 是同时作用于IDO1、IDO2、TDO的多靶点抑制剂。

|
Figure 18 Other IDO1 inhibitors with various scaffolds |
IDO1通过调控色氨酸代谢途径, 影响肿瘤微环境中效应T细胞和免疫耐受调节性T细胞的凋亡与功能, 进而帮助肿瘤细胞逃脱免疫系统的杀伤。因此, IDO1在肿瘤细胞的免疫逃逸中发挥着重要作用, IDO1是一个潜在的肿瘤免疫治疗药物靶标。IDO1蛋白与其抑制剂复合物晶体结构的解析为基于结构的药物分子设计提供了结构基础。IDO1的结合腔主要包括A、B两个疏水性结合腔和A腔底部与血红素中铁离子络合的位点。IDO1蛋白结合口袋存在诱导契合现象, 尤其是B腔随着抑制剂形状和体积的改变, B腔的形状和体积会有较大的改变。这种诱导契合的存在, 既为设计结构多样的小分子抑制剂带来了机遇, 同时也是获得高活性IDO1小分子抑制剂的难点所在。值得关注的是, BMS-986205与不含有亚铁血红素的IDO1结合, 这种结合方式为设计新结构的小分子抑制剂提供了另一种选择。
到目前为止, 已报道了多种结构类型的IDO1小分子抑制剂, 并且有多个候选药物进入临床研究。遗憾的是, 最近报道的IDO1抑制剂的临床试验结果并不理想, 这也提示IDO1抑制剂及其作用机制的研究还有待进一步探索。发现新结构类型、新作用模式的IDO1抑制剂, 既可为肿瘤免疫治疗机制研究提供物质基础, 也可能为靶向IDO1的肿瘤免疫治疗带来新的突破。
| [1] | van Baren N, Van den Eynde BJ. Tryptophan-degrading enzymes in tumoral immune resistance[J]. Front Immunol, 2015, 6: 1–9. |
| [2] | King NJ, Thomas SR. Molecules in focus: indoleamine 2, 3-dioxygenase[J]. Int J Biochem Cell Biol, 2007, 39: 2167–2172. DOI:10.1016/j.biocel.2007.01.004 |
| [3] | Ball HJ, Sanchez-Perez A, Weiser S, et al. Characterization of an indoleamine 2, 3-dioxygenase-like protein found in humans and mice[J]. Gene, 2007, 396: 203–213. DOI:10.1016/j.gene.2007.04.010 |
| [4] | Sugimoto H, Oda S, Otsuki T, et al. Crystal structure of human indoleamine 2, 3-dioxygenase: catalytic mechanism of O2 incorporation by a heme-containing dioxygenase[J]. Proc Natl Acad Sci U S A, 2006, 103: 2611–2616. DOI:10.1073/pnas.0508996103 |
| [5] | Littlejohn TK, Takikawa O, Truscott RJ, et al. Asp274 and his346 are essential for heme binding and catalytic function of human indoleamine 2, 3-dioxygenase[J]. J Biol Chem, 2003, 278: 29525–29531. DOI:10.1074/jbc.M301700200 |
| [6] | Crosignani S, Bingham P, Bottemanne P, et al. Discovery of a novel and selective indoleamine 2, 3-dioxygenase (IDO-1) inhibitor 3-(5-fluoro-1H-indol-3-yl)pyrrolidine-2, 5-dione (EOS200271/ PF-06840003) and its characterization as a potential clinical candidate[J]. J Med Chem, 2017, 60: 9617–9629. DOI:10.1021/acs.jmedchem.7b00974 |
| [7] | Lewis-Ballester A, Pham KN, Batabyal D, et al. Structural insights into substrate and inhibitor binding sites in human indoleamine 2, 3-dioxygenase 1[J]. Nat Commun, 2017, 8: 1693–1701. DOI:10.1038/s41467-017-01725-8 |
| [8] | Tomek P, Palmer BD, Flanagan JU, et al. Discovery and evaluation of inhibitors to the immunosuppressive enzyme indoleamine 2, 3-dioxygenase 1 (IDO1): probing the active site-inhibitor interactions[J]. Eur J Med Chem, 2017, 126: 983–996. DOI:10.1016/j.ejmech.2016.12.029 |
| [9] | Munn DH, Shafizadeh E, Attwood JT, et al. Inhibition of T cell proliferation by macrophage tryptophan catabolism[J]. J Exp Med, 1999, 189: 1363–1372. DOI:10.1084/jem.189.9.1363 |
| [10] | Munn DH, Mellor AL. Indoleamine 2, 3 dioxygenase and metabolic control of immune responses[J]. Trends Immunol, 2013, 34: 137–143. DOI:10.1016/j.it.2012.10.001 |
| [11] | Pietra G, Vitale M, Moretta L, et al. How melanoma cells inactivate NK cells[J]. Oncoimmunology, 2012, 1: 974–975. DOI:10.4161/onci.20405 |
| [12] | Wang D, Saga Y, Mizukami H, et al. Indoleamine-2, 3-dioxygenase, an immunosuppressive enzyme that inhibits natural killer cell function, as a useful target for ovarian cancer therapy[J]. Int J Oncol, 2012, 40: 929–934. DOI:10.3892/ijo.2011.1295 |
| [13] | Fallarino F, Grohmann U, Hwang KW, et al. Modulation of tryptophan catabolism by regulatory T cells[J]. Nat Immunol, 2003, 4: 1206–1212. DOI:10.1038/ni1003 |
| [14] | Hornyak L, Dobos N, Koncz G, et al. The role of indoleamine-2, 3-dioxygenase in cancer development, diagnostics, and therapy[J]. Front Immunol, 2018, 9: 151. DOI:10.3389/fimmu.2018.00151 |
| [15] | Prendergast GC, Malachowski WP, DuHadaway JB, et al. Discovery of IDO1 inhibitors: from bench to bedside[J]. Cancer Res, 2017, 77: 6795–6811. DOI:10.1158/0008-5472.CAN-17-2285 |
| [16] | Cheong JE, Sun L. Targeting the IDO1/TDO2-KYN-AhR pathway for cancer immunotherapy-challenges and opportunities[J]. Trends Pharmacol Sci, 2018, 39: 307–325. DOI:10.1016/j.tips.2017.11.007 |
| [17] | Dinatale BC, Murray IA, Schroeder JC, et al. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling[J]. Toxicol Sci, 2010, 115: 89–97. DOI:10.1093/toxsci/kfq024 |
| [18] | Opitz CA, Litzenburger UM, Sahm F, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor[J]. Nature, 2011, 478: 197–203. DOI:10.1038/nature10491 |
| [19] | Nguyen NT, Kimura A, Nakahama T, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism[J]. Proc Natl Acad Sci U S A, 2010, 107: 19961–19966. DOI:10.1073/pnas.1014465107 |
| [20] | Cheong JE, Ekkati A, Sun L. A patent review of IDO1 inhibitors for cancer[J]. Expert Opin Ther Pat, 2018, 28: 317–330. DOI:10.1080/13543776.2018.1441290 |
| [21] | Weng T, Qiu X, Wang J, et al. Recent discovery of indoleamine-2, 3-dioxygenase 1 inhibitors targeting cancer immunotherapy[J]. Eur J Med Chem, 2018, 143: 656–669. DOI:10.1016/j.ejmech.2017.11.088 |
| [22] | Rohrig UF, Majjigapu SR, Vogel P, et al. Challenges in the discovery of indoleamine 2, 3-dioxygenase 1 (IDO1) inhibitors[J]. J Med Chem, 2015, 58: 9421–9437. DOI:10.1021/acs.jmedchem.5b00326 |
| [23] | Xu YY, Wu JJ. Advances in the development of IDO1 inhibitors[J]. Chin J New Drugs (中国新药杂志), 2016, 25: 425–432. |
| [24] | Cheng YL, Men JX, Zhou JP, et al. Advances in indoleamine 2, 3-dioxygenase 1 inhibitors[J]. J China Pharm Univ (中国药科大学学报), 2017, 48: 361–370. |
| [25] | Du TT, Lai FF, Chen XG. Research progress of indoleamine 2, 3-dioxygenase 1 in tumor immunotherapy[J]. Acta Pharm Sin (药学学报), 2018, 53: 1271–1278. |
| [26] | Yue EW, Douty B, Wayland B, et al. Discovery of potent competitive inhibitors of indoleamine 2, 3-dioxygenase with in vivo pharmacodynamic activity and efficacy in a mouse melanoma model[J]. J Med Chem, 2009, 52: 7364–7367. DOI:10.1021/jm900518f |
| [27] | Yue EW, Sparks R, Polam P, et al. INCB24360 (epacadostat), a highly potent and selective indoleamine-2, 3-dioxygenase 1 (IDO1) inhibitor for immuno-oncology[J]. ACS Med Chem Lett, 2017, 8: 486–491. DOI:10.1021/acsmedchemlett.6b00391 |
| [28] | Wang ZY, Guo W, Zhu JD. Indoleamine-2, 3-dioxygenase inhibitor and preparation method therefor: CN, 089666[P]. 2016-03-24. |
| [29] | Lv HJ, Gui B, Dong Q, et al. Sulfamyl-containing 1, 2, 5-oxadiazole derivative, preparation method therefor and use thereof in pharmaceuticals: CN, 076975[P]. 2016-10-06. |
| [30] | Yang FL, Gui B, Hu QY, et al. Hydroxy amidine derivative, preparation method and use in medicine thereof: CN, 093584[P]. 2017-02-16. |
| [31] | Bartlett MJ, Codelli JA, Corkey BK, et al. Benzimidazole and imadazopyridine carboximidamide compounds: US, 15153527[P]. 2016-11-17. |
| [32] | Paul S, Roy A, Deka SJ, et al. Nitrobenzofurazan derivatives of N'-hydroxyamidines as potent inhibitors of indoleamine-2, 3-dioxygenase 1[J]. Eur J Med Chem, 2016, 121: 364–375. DOI:10.1016/j.ejmech.2016.05.061 |
| [33] | Jaen JC, Osipov M, Powers JP, et al. Immunoregulatory agents: US, 034449[P]. 2015-12-10. |
| [34] | Beck HP, Jaen JC, Osipov M, et al. Immunoregulatory agents: US, 059311[P]. 2016-05-12. |
| [35] | Beck HP, Jaen JC, Osipov M, et al. Immunoregulatory agents: US, 059316[P]. 2016-05-12. |
| [36] | Beck HP, Jaen JC, Osipov M, et al. Immunoregulatory agents: US, 059271[P]. 2016-05-12. |
| [37] | Nelp MT, Kates PA, Hunt JT, et al. Immune-modulating enzyme indoleamine 2, 3-dioxygenase is effectively inhibited by targeting its apo-form[J]. Proc Natl Acad Sci U S A, 2018, 115: 3249–3254. DOI:10.1073/pnas.1719190115 |
| [38] | Cady SG, Sono M. 1-Methy-DL-typtophan, β-(3-benzofuranyl)-DL-alanine (the oxygen analog of tryptophan), and β-[3-benzo (b)thienyl]-DL-alanine (the sulfur analog of tryptophan) are competitive inhibiors for indoleamine 2, 3-dioxygenase[J]. Arch Biochem Biophys, 1991, 291: 326–333. DOI:10.1016/0003-9861(91)90142-6 |
| [39] | Muller AJ, Malachowski WP, Prendergast GC. Indoleamine 2, 3-dioxygenase in cancer: targeting pathological immune tolerance with small-molecule inhibitors[J]. Expert Opin Ther Targets, 2005, 9: 831–849. DOI:10.1517/14728222.9.4.831 |
| [40] | Kudo Y, Boyd C. The role of l-tryptophan transport in l-tryptophan degradation by indoleamine 2, 3-dioxygenase in human placental explants[J]. J Physiol, 2001, 531: 417–423. DOI:10.1111/tjp.2001.531.issue-2 |
| [41] | Metz R, Rust S, Duhadaway JB, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: a novel IDO effector pathway targeted by D-1-methyl-tryptophan[J]. Oncoimmunology, 2012, 1: 1460–1468. DOI:10.4161/onci.21716 |
| [42] | Fu T, He Q, Sharma P. The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti-CTLA-4 therapy[J]. Cancer Res, 2011, 71: 5445–5454. DOI:10.1158/0008-5472.CAN-11-1138 |
| [43] | Xie DL, Wu J, Lou YL, et al. Tumor suppressor TSC1 is critical for T-cell anergy[J]. Proc Natl Acad Sci U S A, 2012, 109: 14152–14157. DOI:10.1073/pnas.1119744109 |
| [44] | Colwell J. Indoximod combo triggers responses in melanoma[J]. Cancer Discov, 2017, 7: 542–543. |
| [45] | Caspari P, Banerjee T, Malachowski WP, et al. Structure-activity study of brassinin derivatives as indoleamine 2, 3-dioxygenase inhibitors[J]. J Med Chem, 2006, 49: 684–692. DOI:10.1021/jm0508888 |
| [46] | Muller AJ, DuHadaway JB, Donover PS, et al. Inhibition of indoleamine 2, 3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy[J]. Nat Med, 2005, 11: 312–319. DOI:10.1038/nm1196 |
| [47] | Tanaka M, Li X, Hikawa H, et al. Synthesis and biological evaluation of novel tryptoline derivatives as indoleamine 2, 3-dioxygenase (IDO) inhibitors[J]. Bioorg Med Chem, 2013, 21: 1159–1165. DOI:10.1016/j.bmc.2012.12.028 |
| [48] | Dolusic E, Larrieu P, Blanc S, et al. Indol-2-yl ethanones as novel indoleamine 2, 3-dioxygenase (IDO) inhibitors[J]. Bioorg Med Chem, 2011, 19: 1550–1561. DOI:10.1016/j.bmc.2010.12.032 |
| [49] | Dolusic E, Larrieu P, Blanc S, et al. Discovery and preliminary SARs of keto-indoles as novel indoleamine 2, 3-dioxygenase (IDO) inhibitors[J]. Eur J Med Chem, 2011, 46: 3058–3065. DOI:10.1016/j.ejmech.2011.02.049 |
| [50] | Coluccia A, Passacantilli S, Famiglini V, et al. New inhibitors of indoleamine 2, 3-dioxygenase 1: molecular modeling studies, synthesis, and biological evaluation[J]. J Med Chem, 2016, 59: 9760–9773. DOI:10.1021/acs.jmedchem.6b00718 |
| [51] | Qian S, He T, Wang W, et al. Discovery and preliminary structure-activity relationship of 1H-indazoles with promising indoleamine-2, 3-dioxygenase 1 (IDO1) inhibition properties[J]. Bioorg Med Chem, 2016, 24: 6194–6205. DOI:10.1016/j.bmc.2016.10.003 |
| [52] | Qian S, Wang ZY, Yang LL, et al. 1H-indazole derivative and use thereof as IDO inhibitor: CN, 099845[P]. 2017-08-10. |
| [53] | Wang H, Zhang G, Guo Y, et al. Novel 5 or 8-substituted imidazo[1, 5-a]pyridines as indoleamine and/or tryptophane 2, 3-dioxygenases: US, 078787[P]. 2016-10-13. |
| [54] | Kumar S, Jaller D, Patel B, et al. Structure based development of phenylimidazole-derived inhibitors of indoleamine 2, 3-dioxygenase[J]. J Med Chem, 2008, 51: 4968–4977. DOI:10.1021/jm800512z |
| [55] | Mautino MR, Kumar S, Jaipuri F, et al. IDO inhibitors: US, 041609[P]. 2009-10-29. |
| [56] | Huang Q, Zheng M, Yang S, et al. Structure-activity relationship and enzyme kinetic studies on 4-aryl-1H-1, 2, 3-triazoles as indoleamine 2, 3-dioxygenase (IDO) inhibitors[J]. Eur J Med Chem, 2011, 46: 5680–5687. DOI:10.1016/j.ejmech.2011.08.044 |
| [57] | Rohrig UF, Majjigapu SR, Grosdidier A, et al. Rational design of 4-aryl-1, 2, 3-triazoles for indoleamine 2, 3-dioxygenase 1 inhibition[J]. J Med Chem, 2012, 55: 5270–5290. DOI:10.1021/jm300260v |
| [58] | Mautino MR, Kumar S, Jaipuri F, et al. Imidazole derivatives as IDO inhibitors: US, 054289[P]. 2011-05-12. |
| [59] | Mautino M, Kumar S, Waldo J, et al. Fused imidazole derivatives useful as IDO inhibitors: US, 033245[P]. 2012-10-18. |
| [60] | Mautino MR, Jaipuri FA, Waldo J, et al. NLG919, a novel indoleamine-2, 3-dioxygenase (IDO)-pathway inhibitor drug candidate for cancer therapy[J]. Cancer Res, 2013, 73: 491. DOI:10.1158/1538-7445.AM2013-491 |
| [61] | Peng YH, Ueng SH, Tseng CT, et al. Important hydrogen bond networks in indoleamine 2, 3-dioxygenase 1 (IDO1) inhibitor design revealed by crystal structures of imidazoleisoindole derivatives with IDO1[J]. J Med Chem, 2016, 59: 282–293. DOI:10.1021/acs.jmedchem.5b01390 |
| [62] | Li Q, Gao D. Fused-ring compounds, pharmaceutical composition and uses thereof: CN, 073260[P]. 2016-08-25. |
| [63] | Zhang H, Liu S. Heterocycles useful as IDO and TDO inhibitors: CN, 079104[P]. 2016-10-20. |
| [64] | Armer R, Bingham M, Pesnot T, et al. 4H-Imidazo[1, 5-a] indole derivatives and their use as indoleamine 2, 3-dioxygenase (IDO) and/or tryptophan 2, 3-dioxygenase (TDO2) modulators: GB, 052868[P]. 2016-04-07. |
| [65] | Sherer BA. Cyclohexyl-ethyl substituted diaza- and triaza-tricyclic compounds as indole-amine-2, 3-dioxygenase (IDO) antagonists for the treatment of cancer: US, 048474[P]. 2016-03-10. |
| [66] | Liu SL, Wang DH, Liang GB, et al. Tricyclic compound serving as immunomodulator: CN, 074141[P]. 2017-08-24. |
| [67] | Tu WY, Xu GJ, Zhang HT, et al. Imidazo isoindole derivative, preparation method therefor and medical use thereof: CN, 079054[P]. 2016-10-27. |
| [68] | Alexandre JAC, Swan MK, Latchem MJ, et al. New 4-amino-1, 2, 3-triazole inhibitors of indoleamine 2, 3-dioxygenase form a long-lived complex with the enzyme and display exquisite cellular potency[J]. Chembiochem, 2018, 19: 552–561. DOI:10.1002/cbic.201700560 |
| [69] | Tojo S, Kohno T, Tanaka T, et al. Crystal structures and structure-activity relationships of imidazothiazole derivatives as IDO1 inhibitors[J]. ACS Med Chem Lett, 2014, 5: 1119–1123. DOI:10.1021/ml500247w |
| [70] | Cheng MF, Hung MS, Song JS, et al. Discovery and structure-activity relationships of phenyl benzenesulfonylhydrazides as novel indoleamine 2, 3-dioxygenase inhibitors[J]. Bioorg Med Chem Lett, 2014, 24: 3403–3406. DOI:10.1016/j.bmcl.2014.05.084 |
| [71] | Lin SY, Yeh TK, Kuo CC, et al. Phenyl benzenesulfonylhydrazides exhibit selective indoleamine 2, 3-dioxygenase inhibittion with potent in vivo pharmacodynamic activity and antitumor efficacy[J]. J Med Chem, 2016, 59: 419–430. DOI:10.1021/acs.jmedchem.5b01640 |
| [72] | Gao D, Li Y. Identification and preliminary structure-activity relationships of 1-indanone derivatives as novel indoleamine-2, 3-dioxygenase 1 (IDO1) inhibitors[J]. Bioorg Med Chem, 2017, 25: 3780–3791. DOI:10.1016/j.bmc.2017.05.017 |
| [73] | Williams DE, Steino A, de Voogd NJ, et al. Halicloic acids A and B isolated from the marine sponge Haliclona sp. collected in the Philippines inhibit indoleamine 2, 3-dioxygenase[J]. J Nat Prod, 2012, 75: 1451–1458. DOI:10.1021/np300345j |
| [74] | Terentis AC, Freewan M, Sempertegui Plaza TS, et al. The selenazal drug ebselen potently inhibits indoleamine 2, 3-dioxygenase by targeting enzyme cysteine residues[J]. Biochemistry, 2010, 49: 591–600. DOI:10.1021/bi901546e |
| [75] | Banerjee M, Middya S, Shrivastava R, et al. Aminonitriles as kynurenine pathway inhibitors: IB, 059705[P]. 2014-09-18. |
| [76] | Banerjee M, Middya S, Shrivastava R, et al. Inhibitors of the kynurenine pathway: US, 024920 [P]. 2014-11-20. |