 2018, Vol. 53
2018, Vol. 53



2. 中国药科大学药学院, 江苏 南京 210009;
3. 天士力控股集团有限公司研究院化学药品开发中心, 天津 300410
2. College of Pharmacy, China Pharmaceutical University, Nanjing 210009, China;
3. Chemical Medicine R & D Center, Tasly Academy, Tianjin Tasly Holding Group Co., Ltd., Tianjin 300410, China
肝纤维化是一个由慢性肝病转化而来的严重健康问题, 若未得到合理的治疗将发展成肝硬化、肝衰竭或肝癌, 最终导致死亡。目前针对肝纤维化的治疗局限于控制病因或中药治疗, 至今仍然没有进入临床应用的化学药物[1]。肝纤维化属于可逆性病变, 若进行积极有效的治疗, 可以减轻或逆转。随着人们对肝纤维化发生、发展机制取得突破性的认知, 已有多种在研药物通过干预肝纤维化过程中的重要环节, 在临床前动物模型研究和/或临床研究中取得了良好的抗肝纤维化效果。
1 肝纤维化流行现状肝纤维化是慢性肝病在修复过程中导致的病理状态, 慢性肝病主要包括慢性乙型肝炎(chronic hepatitis B, CHB)、慢性丙型肝炎(chronic hepatitis C, CHC)、酒精性肝病(alcoholic liver disease, ALD)和非酒精性脂肪性肝炎(non-alcoholic steatohepatitis, NASH)等, 严重危害人类健康。慢性肝病导致的肝纤维化是肝硬化的早期阶段, 肝硬化病程超过5年, 约10%的患者将发展为肝癌。截止2015年, 流行病学调查结果显示, 世界范围内约有乙型肝炎病毒(hepatitis B virus, HBV)携带者2.57亿人, 88.7万人死于HBV导致的肝硬化; 丙型肝炎病毒(hepatitis C virus, HCV)携带者约7 100万人, 39.9万人死于CHC导致的肝硬化[2]。肝硬化每年造成全世界120万人死亡, 死亡率大于5种主要癌症, 2015年世界因肝癌死亡人数约81万[3]。在全球范围内中国肝病负担最重, 约3亿人受到影响[4], 2015年调查结果显示, 肝癌更是造成中国约42.2万人死亡[5]。
2 肝纤维化发病机制肝纤维化是由多种病因学因素引起, 如酗酒、病毒性肝炎、自身免疫性肝炎、非酒精性脂肪性肝炎、原发胆汁性肝硬化和原发胆管性肝硬化[6]。ALD是指长期大量饮酒所致的一种肝脏疾病, 过度饮酒会导致酒精性脂肪肝, 戒酒可逆转这一症状[7]。慢性病毒性肝炎是由肝脏病毒引起的, 其治疗主要通过抑制病毒复制和抗病毒, 从而使症状得到改善。HBV多用恩替卡韦或替诺福韦进行治疗[8], HCV可以用索非布韦或利巴韦林进行治疗[9]。NASH导致的纤维化状况, 要通过控制饮食、进行运动、控制体重等进行控制治疗。
肝纤维化是持续性肝损伤的必然结果, 持续性肝损伤的成因与多种信号因子有关, 包括趋化因子信号、脂肪因子信号、神经内分泌信号、血管生成信号及NADPH氧化酶信号等。肝脏病毒、损伤、过量脂肪酸或胆汁酸等可以诱导肝细胞发生炎症反应。炎症反应发生时会刺激Kupffer细胞等释放转化生长因子-β1 (transforming growth factor-β1, TGF-β1)、结缔组织生长因子(connective tissue growth factor, CTGF)、血小板衍生生长因子(platelet-derived growth factor, PDGF)、血管内皮生长因子(vascular endothelial growth factor, VEGF)、纤维母细胞生长因子(fibroblast growth factor, FGF)及角质形成细胞生长因子, 这些生长因子将导致肝星状细胞(hepatic stellate cell, HSC)或肌成纤维细胞(myofibroblasts, MFB)活化、分化与增殖, 合成大量以胶原为主要成分的细胞外基质(extracellular matrix, ECM), 使之逐渐沉积从而引起肝纤维化[10]。
3 在研药物和靶点大多数在研药物的主要作用机制与抑制肝纤维化形成过程中的各种因素有关, 主要包括HSC活化增殖、炎症、氧化应激及ECM生成等。
3.1 抑制HSC活化增殖HSC活化是肝纤维化形成的关键环节, 被视为抗肝纤维化药物重要的靶标[11]。HSC活化增殖过程与一系列复杂的细胞因子介导的信号通路有关, 如TGF-β、PDGF、FGF等, 且这些通路并非互相独立, 而是彼此联系、互相影响、共同发挥作用。一些小分子酶同样也可以促进HSC活化, 如组织蛋白酶B (cathepsin B)、羟甲基戊二酰辅酶还原酶(HMG-CoA还原酶)等。若对上述细胞通路或信号因子进行阻断, 可有效抑制HSC活化增殖, 使肝纤维化状况得到改善[12]。现将抑制HSC活化增殖相关在研药物汇总至表 1。
| Table 1 Anti-hepatic fibrosis drugs relating to inhibiting activation and proliferation of hepatic stellate cell (HSC) |
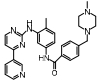
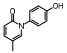
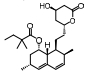
TGF-β1是目前发现的最强的促纤维化细胞因子, 可以诱导HSC活化, 促进ECM分泌, 形成肝纤维化[13]。Smad蛋白家族是TGF-β1关键的下游信号, 包括膜受体激活Smads (Smad1、2、3、5、8)、通用Smad (Smad4)及抑制性Smads (Smad6、7)三大类[14], Smad7可抑制Smad3和Smad4的活性而阻断TGF-β信号效应[15]。
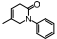
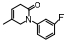
吡非尼酮(pirfenidone, PFD)是一种广谱抗纤维化药物, 其抗肝纤维化研究已进入Ⅱ期临床试验阶段。对34名受试者进行为期2年的临床研究, 结果显示, 67%的患者纤维化状况出现缓解, 改善了患者的炎症、纤维化和脂肪变性, 降低了TGF-β1水平, 减少了纤维化受体的表达[16]。氟非尼酮(fluorofenidone, AKF-PD)是PFD的me-better药物, AKF-PD在四氯化碳、二甲基亚硝胺以及猪血清诱导的大鼠肝纤维化模型中均有显著的抗纤维化作用[17]。马洛替酯(malotilate)可以显著减轻二甲基亚硝胺诱导的实验性大鼠的肝损伤, 使肝纤维化程度得到改善, Smad3/4表达水平下调, Smad7表达水平上调[15]。
3.1.2 PDGFPDGF是HSC分化过程中重要的有丝分裂原, 主要来源于免疫细胞、胆管细胞、血小板等。PDGFs与细胞表面的PDGF-α/β受体结合成二聚体, 通过下游酪氨酸激酶磷酸化作用于细胞内受体, 使活化的HSC增殖分裂(图 1)[18], 因此抑制PDGF受体有助于抑制活化HSC的功能[19]。

|
Figure 1 The involvement of platelet-derived growth factor (PDGF) in liver fibrosis process. PDGFR: Platelet-derived growth factor receptor; MFB: Myofibroblasts |
Sorafenib能抑制血管内皮生长因子受体2 (vas cular endothelial growth factor receptor 2, VEGFR-2)和PDGF-β受体, 明显改善肝损伤和肝纤维化, 促进血管生成[20]。在肝硬化临床试验中, sorafenib有显著肝脏保护作用, 但Child-Puge B患者治疗期间出现肝硬化恶化情况生存率低于Child-Puge A患者, 毒性分布无明显区别[21]。Sorafenib未来有可能成为一种新型的治疗肝纤维化的方法, 为一些慢性肝病提供治疗选择, 但其有效剂量及不良反应尚需进一步确认[22]。Imatinib mesylate是PDGF-α/β受体的抑制剂。在胆汁性大鼠肝纤维化模型中, imatinib短期内可以明显降低HSC的增殖, 在肝纤维化早期有效, 而对长期肝损伤效果不明显[23]。
3.1.3 FGFFGF家族有20多种成员, 分为典型FGFs、内分泌FGFs和细胞内FGFs, 与4种不同配体(FGFR1~4)结合, 促进早期胚胎发展、器官形成, 对于成年组织可以调节新陈代谢、修复组织再生、激活信号通路。FGFR1介导的信号传导与肝纤维化、肝硬化密切相关; 内分泌FGFs中FGF15/19和FGF21可以调控HSC, 抑制肝纤维化发生, 在慢性酒精小鼠肝纤维化模型中FGF21主要作用于改善肝脏脂质堆积和肝脏炎症损伤[24]。
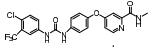
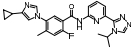
BMS-986036是一种聚乙二醇修饰的FGF21类似物, Ⅱ期临床研究对74名NASH患者进行为期16周的双盲对照试验, 皮下注射BMS-986036或空白安慰剂, BMS-986036组相对于安慰组显著降低NASH患者的肝脂肪含量, 结果表明BMS-986036可以改善NASH、脂肪变性、肝损伤和纤维化状况, 但受试者出现轻微不良反应, 如腹泻、恶心、排便频繁, 未发生严重不良反应[25]。FGFR1抑制剂hydronidone可以改善四氯化碳、二甲基亚硝胺和人血清白蛋白在大鼠和小鼠模型中诱发的肝纤维化状况, Ⅰ期临床研究结果显示药物吸收速率较快, 受试者有良好的依从性, 无明显的不良反应, 但食物摄取会降低hydronidone的吸收速率[26]。
3.1.4 HMG-CoA还原酶他汀类代表药物辛伐他汀(simvastatin)为HMG-CoA还原酶抑制剂, 在高脂饲养导致的非酒精性肝纤维化大鼠模型中可显著增加AMP依赖的蛋白激酶(AMPK)活性, 抑制HSC活化, 减轻肝纤维化状况, 逆转肝脏继发的纤维化改变[27]。
3.1.5 Cathepsin BCathepsin B参与HSC的增殖分化, 抑制cathepsin B的活性可以减缓或抑制HSC的增殖[28]。VBY-376是一种cathepsin B抑制剂, 可以缓解NASH患者的肝纤维化状况。在肝脏毒素引起的小鼠纤维化模型中, 使用VBY-376分别进行预防和逆转治疗, 结果显示VBY-376可以明显减轻纤维化状况, 但预防治疗效果更佳, 已在46名健康志愿者中完成Ⅰ期临床试验并取得了理想的结果[29]。
3.2 抑制炎症细胞毒素、肝脏损伤及脂肪过度积聚等可以使肝脏发生炎症反应, 炎症细胞释放的炎症因子导致免疫细胞旁分泌, 从而促进HSC活化而引起肝纤维化, 同时释放细胞凋亡因子使炎症反应扩大。通过抑制肝内脂肪积聚、促炎因子释放及免疫细胞旁分泌可以有效控制肝纤维化。促炎因子主要包括趋化因子、免疫细胞旁分泌细胞凋亡因子、半乳凝素-3 (galectin-3, Gal-3)和过氧化物酶体增殖物等。炎症导致肝纤维化过程如图 2[30]。与抑制炎症相关药物汇总至表 2。

|
Figure 2 The process of inflammation leading to hepatic fibrosis. CCR2/5: CC chemokine receptor types 2/5; CCL2: CC chemokine ligand type 2; FXR: Farnesoid-X receptor; ACC: Acetyl CoA carboxylase; GLP-1: Glucagon-like peptide-1; Gal-3: Galectin-3; ASK1: Apoptosis-signal-regulating kinase 1; TNF-α: Tumor necrosis factor-α; PPAR: Peroxisome proliferators-activated receptor |
| Table 2 Anti-hepatic fibrosis drugs relating to inhibiting inflammation |
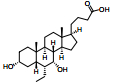
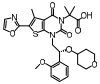
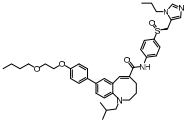
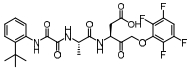
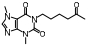
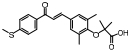
脂肪酸在肝内过多积聚产生肝脏毒性损伤作用, 导致肝脏细胞发生炎症反应。胆汁酸激活的法尼酯X受体(farnesoid-X receptor, FXR)信号可以增强胰岛素敏感性, 减少肝细胞糖质和脂肪新生[31]。FXR激动剂obeticholic acid (OCA, INT-747)是一种半合成的鹅去氧胆酸, 在283名NASH患者中进行为期72周的双盲随机安慰剂对照实验, 结果显示用药组纤维化状况有明显改善, 仅少数患者出现瘙痒等轻微不良反应[32]。乙酰辅酶A羧化酶(acetyl CoA carboxylase, ACC)是催化肝脏内脂肪从头合成第一步的限速酶, 抑制ACC可以减少肝脏内脂肪的积累。GS-0976作为一种ACC抑制剂, 在Ⅱ期随机双盲对照临床研究中, 对126例NASH并伴有纤维化患者进行为期12周临床试验, 结果显示用药组可以使肝脏纤维化标志物TIMP-1明显降低, 但未能改变肝硬度[33]。胰高血糖素样肽-1 (glucagon-like peptide-1, GLP-1)通过各种途径增加胰岛素释放和降低胰高血糖素分泌, 减少肝脏脂肪变性, 可直接改善肝纤维化状态。利拉鲁肽(liraglutide)是一种GLP-1类似物, 在随机双盲安慰剂对照的Ⅱ期多中心临床试验中, 对患者进行了为期48周的研究, 肝脏活检结果显示, 用药组39%的受试者NASH状况得到改善, 肝纤维化程度未出现进一步加重, 安慰剂组仅有9%受试者出现好转[34]。
3.2.2 趋化因子趋化因子是一类具有化学趋化作用的小分子蛋白质, 有调控炎症反应的功能。为了应对肝脏炎症, Kupffer细胞分泌CC趋化因子配体2/5 (CC chemokine ligand type 2/5, CCL2/5), 与相应的CC趋化因子受体2/5 (CC chemokine receptor types 2/5, CCR2/5)结合, 这些促炎因子使炎症反应扩大, 导致纤维化状况严重[30]。
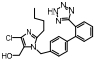
Cenicriviroc是CCR2和CCR5的双重抑制剂, 在Ⅱ期临床研究中, 30名轻中度肝损伤受试者结果显示cenicriviroc可以快速阻断CCR2和CCR5, 150 mg cenicriviroc可以用于治疗轻中度肝损伤, 且仅有1名患者出现口干、肠胃胀气等轻微不良反应, 但cenicriviroc对重度肝损伤及NASH患者的疗效还需进一步验证[35]。
3.2.3 细胞凋亡因子细胞凋亡因子有Pan-caspase、肿瘤坏死因子(TNF-α)、细胞凋亡信号调节激酶1 (apoptosis-signal-regulating kinase 1, ASK1)等。Pan- caspase通过激活凋亡蛋白酶促进细胞凋亡; TNF-α参与凋亡配体激活和表达; ASK1可以被促纤维化刺激因子活化, 激活诱导细胞凋亡的信号通道, 导致细胞凋亡。
Emricasan可以抑制caspase活性, 减少细胞凋亡, 改善炎症环境, 抑制HSC活化[36], 被用于NASH患者的肝纤维化治疗。在多中心的Ⅱb期临床试验中, 对74例肝硬化患者进行为期3个月的研究结果表明, emricasan可以改善重度肝硬化患者的肝功能, 且安全性耐受性良好, 不良反应出现情况与安慰剂组相似[37]。TNF-α抑制剂pentoxifylline是一种甲基黄嘌呤的衍生物, 可以通过抑制TNF-α基因转录降低炎症状态, 减少氧化应激, 改善NASH患者肝纤维化程度, 但是对酒精性肝炎患者无明显作用[38]。ASK1抑制剂selonsertib (GS-4997)在74例NASH患者中进行了为期24周的临床研究, 结果显示降低了细胞凋亡过程中的生物标志物, 改善了NASH患者2~3期的纤维化状况, 目前处于临床Ⅲ期研究中[39]。
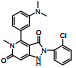
3.2.4 过氧化物酶体增殖物激活受体(peroxisome proliferators-activated receptors, PPARs)PPARs是核受体的一类, 包括PPAR-α、PPAR-β/δ、PPAR-γ。激活后的PPAR可以抑制Kupffer细胞释放炎症因子, 减轻炎症反应。Elafibranor (GFT-505)是一种PPAR-α/δ激动剂, 在双盲对照Ⅱ期临床试验中, 276例NASH患者显示elafibranor可以改善NASH状况, 缓解脂肪性肝炎, 有明显的肝保护作用和良好的耐受性, 对照组间的不良反应发生率基本一致[40]。Elafibranor治疗伴有轻度肝纤维化的NASH患者也已进入FDA的快速通道。
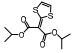
3.2.5 Gal-3Gal-3是一种具有免疫作用的凝集素, 过度表达能够明显促进炎症反应, 使肝纤维化进一步扩大[41]。炎症状态下, Gal-3在Kupffer细胞及免疫细胞中大量表达。Gal-3抑制剂GM-CT-01和GR- MD-02均能降低肝脏纤维化状况。GR-MD-02是一种复杂的碳水化合物, 在对肝纤维化的NASH患者进行有效性评估发现, 肝纤维化及肝脏僵硬程度得到明显改善, 且安全性耐受性良好, 其临床Ⅱ期试验正在进行之中[42]。
3.3 抑制氧化应激氧化应激是纤维化的诱发因素之一, 减少氧化损伤可改善肝功能和降低纤维化发生。减少氧化损伤可以通过抑制NADPH氧化酶(NADPH-oxidase, NOX)或血管紧张素受体等来实现。抑制氧化应激相关在研药物基本信息见表 3。
| Table 3 Anti-hepatic fibrosis drugs relating to inhibiting oxidative stress |
NOX是一种多组分跨膜酶复合物, 其成员包括NOX1、NOX3、NOX4、NOX5、DUOX1和DUOX2。在肝纤维化过程中, NOX发挥重要作用。在肝脏中, Kupffer细胞、巨噬细胞和其他免疫细胞表达NOX2, 而肝细胞和内皮细胞表达NOX1、NOX2和NOX4。NOX1、NOX2和NOX4介导不同的配体刺激, 促进纤维化, 主要表现为促进细胞增殖、迁移、ECM合成、重塑、炎症以及增强收缩性[31]。在四氯化碳或胆道结扎形成的肝纤维化小鼠模型中, GKT137831通过抑制NOX1和NOX4, 减少了肝纤维化程度[43]。
3.3.2 血管紧张素Ⅱ (angiotensinⅡ, AngⅡ)AngⅡ通过刺激非吞噬细胞NOX诱导氧化应激来促进纤维化。Losartan是一种AngⅡ受体阻滞剂。对14例慢性丙肝肝纤维化患者进行为期18个月的临床试验, 结果显示50%受试者炎症、纤维化状况有所改善[44]。
3.4 抑制ECM生成ECM主要由Ⅰ、Ⅲ型胶原蛋白组成, 正常情况下ECM生成与沉降处于相对平衡状态, 发生肝损伤时, 组成ECM的胶原蛋白过量产生且发生交联, 交联后不易被蛋白酶降解, 因此抑制ECM产生或交联是一种有效逆转肝纤维化的方法。与此相关的酶主要有基质金属蛋白酶(matrix degrading metalloproteinases, MMPs)和赖氨酰氧化酶(LOX)等。抑制ECM生成相关抗肝纤维化药物见表 4。
| Table 4 Anti-hepatic fibrosis drugs relating to inhibiting generation and degradation of extracellular matrix |
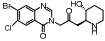
正常情况下ECM可以被MMPs降解; 在各种肝损伤的刺激下, 活化的MMPs被组织金属蛋白抑制因子(tissue inhibitors of metalloproteinases, TIMPs)抑制, ECM在肝细胞内过度沉积, 导致肝纤维化[45]。Halofuginone是喹诺酮类小分子衍生物, 可以特异性抑制I型前胶原合成, 在四氯化碳肝硬化大鼠模型中可抑制I型前胶原的表达与合成, 大幅度降低TIMP水平, 被视为一个有前途的抗肝纤维化药物[46]。
3.4.2 LOXLOX家族对胶原蛋白交联有明显作用, 对硫代乙酰胺诱导的肝纤维化小鼠给予LOX家族中LOXL2的抑制剂可以明显抑制胶原蛋白交联[47]。Simtuzumab (GS-6624)是一种LOXL2的单克隆抗体, 对NASH诱发的肝硬化和纤维化有良好的治疗效果。在Ⅱa期临床试验中, 对18名晚期纤维化患者进行为期22周的研究, 结果显示受试者有良好的耐受性, 目前正在进行Ⅱb期临床试验[48]。
4 联合用药对单一环节、单一靶点进行阻断或抑制已显示出明显的抗纤维化活性, 但是由于肝纤维化的成因涉及多因子、多途径、多细胞, 所以研究多靶点多种药物联合作用以期达到更优的效果。
4.1 GLP-1R和FXRFXR抑制剂OCA可以降低NASH小鼠的脂肪含量, GLP-1R激动剂IP-118既可以降低脂肪含量, 也可以降低ALT、AST。在饮食诱导的NASH及肥胖的小鼠模型中, 两者联合用药比单一使用IP-118能明显降低体重, 进一步提高葡萄糖耐受性, 降低肝脏脂肪含量[49]。
4.2 天然药物联合用药表没食子儿茶素没食子酸酯、牛磺酸和三羟基异黄酮是3种从天然产物中提取的具有不同抗纤维化作用的化合物, 在四氯化碳肝纤维化大鼠模型中使用上述3种化合物联合给药, 结果显示治疗肝纤维化的效果优于单一用药[50], 其机制可能与加快自由基清除, 减轻脂质过氧化损伤, 同时抑制TGF-β1的表达, 减少细胞外基质的合成有关[51]。
5 结语与展望不同类型肝纤维化成因不同, 随着对发病机制的研究越来越深入, 人们对肝纤维化治疗药物作用靶点的认识也越来越清晰, 除了文章中所陈述的这些靶点, 还有COX-2酶抑制剂、低氧诱导因子1α (hypoxia-inducible factor-1alpha, HIF-1α)、Gas6/Axl等。许多药物已在研发过程中显示明确的抗肝纤维化效果, 为肝纤维化治愈提供了极大的可能性, 但是单一药物往往会因为靶点单一、给药剂量大等因素导致明显的不良反应。在这种情况下, 联合用药在临床研究中显示出了提高疗效并降低毒副作用的优势, 因此针对多靶点的联合用药是抗肝纤维化药物研发的一个重要方向。此外, 还有许多生物类药物取得十分瞩目的进展, 如serelaxin, 基因治疗也是未来抗肝纤维化不可或缺的研究方向。
| [1] | Sun M, Kisseleva T. Reversibility of liver fibrosis[J]. Clin Res Hepatol Gas, 2015, 39: S60–S63. DOI:10.1016/j.clinre.2015.06.015 |
| [2] | World Health Organization. Global hepatitis report 2017[R]. Geneva: WHO. 2017. |
| [3] | Fitzmaurice C, Allen C, Barber RM, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015:a systematic analysis for the global burden of disease study[J]. JAMA Oncol, 2017, 3: 524–548. DOI:10.1001/jamaoncol.2016.5688 |
| [4] | Wang FS, Fan JG, Zhang Z, et al. The global burden of liver disease:the major impact of China[J]. Hepatology, 2014, 60: 2099–2108. DOI:10.1002/hep.v60.6 |
| [5] | Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015[J]. CA Cancer J Clin, 2016, 66: 115–132. DOI:10.3322/caac.21338 |
| [6] | Bataller R, Brenner DA. Liver fibrosis[J]. J Clin Invest, 2005, 115: 209–218. DOI:10.1172/JCI24282 |
| [7] | Lieber CS, Jones DP, Decarli LM. Effects of prolonged ethanol intake:production of fatty liver despite adequate diets[J]. J Clin Invest, 1965, 44: 1009–1021. DOI:10.1172/JCI105200 |
| [8] | Wang GQ, Wang FS, Cheng J, et al. The guideline of prevention and treatment for chronic hepatitis B:a 2015 update[J]. Chin J Clin Hepatol (临床肝胆病杂志), 2015, 31: 1941–1960. |
| [9] | Mangia A, Susser S, Piazzolla V, et al. Sofosbuvir and ribavirin for genotype 2 HCV infected patients with cirrhosis:a real life experience[J]. J Hepatol, 2017, 66: 711–717. DOI:10.1016/j.jhep.2016.12.002 |
| [10] | Fu LZ, Zhen T, Zhang YS. Advances in understanding the role of TGF-β/Smad signalling pathways in the pathogenesis of livers fibrosis[J]. Chin J Clin Pharm Therap (中国临床药理与治疗学), 2014, 19: 1189–1195. |
| [11] | Karthikeyan S, Potter JJ, Geschwind JF, et al. Deregulation of energy metabolism promotes antifibrotic effects in human hepatic stellate cells and prevents liver fibrosis in a mouse model[J]. Biochem Biophs Res Commun, 2015, 469: 463–469. |
| [12] | Yu H, Xin D, Jian L. Modulation of hepatic stellate cells and reversibility of hepatic fibrosis[J]. Exp Cell Res, 2017, 352: 420–426. DOI:10.1016/j.yexcr.2017.02.038 |
| [13] | Xu MC, Zhang F, ZhuGe YZ. Current research status of mechanisms of the development and progression of liver fibrosis[J]. Chin J Clin Hepatol (临床肝胆病杂志), 2016, 32: 1806–1809. |
| [14] | Zhao SS, Shao RG, He HW. Potential targets for anti-liver fibrosis[J]. Acta Pharm Sin (药学学报), 2014, 49: 1365–1371. |
| [15] | Huang H, Kang Y, Huang X. Effect of malotilate on expression of Smads protein in rats with dimenthylnitrosamine-induced hepatic fibrosis[J]. J Prac Hepatol (实用肝脏病杂志), 2015, 18: 525–529. |
| [16] | Florescontreras L, Sandovalrodríguez AS, Menaenriquez MG, et al. Treatment with pirfenidone for two years decreases fibrosis, cytokine levels and enhances CB2 gene expression in patients with chronic hepatitis C[J]. BMC Gastroenterol, 2014, 14: 131–141. DOI:10.1186/1471-230X-14-131 |
| [17] | Huang JM. Observation of the Effect of Fluorofenidone on Rat Hepatic Stellste Cell Proliferation (氟非尼酮对大鼠肝星状细胞增殖作用的观察)[D]. Changsha: Central South University, 2012. |
| [18] | Dobie R, Connelly J, Henderson NC. PDGF-mediated regulation of liver fibrosis[J]. Curr Pathobiol Rep, 2015, 3: 225–233. DOI:10.1007/s40139-015-0096-9 |
| [19] | Wu XX, Zhang CZ, Wang X, et al. Targeting angiogenesis and vascular remodeling as a novel therapeutic approach to liver fibrosis[J]. Acta Pharm Sin (药学学报), 2015, 50: 535–540. |
| [20] | Borkham-Kamphorst E, Weiskirchen R. The PDGF system and its antagonists in liver fibrosis[J]. Cytokine Growth Factor Rev, 2016, 28: 53–61. DOI:10.1016/j.cytogfr.2015.10.002 |
| [21] | DA Fonseca LG, Barroso-sousa R, Bento AD, et al. Safety and efficacy of sorafenib in patients with Child-Pugh B advanced hepatocellular carcinoma[J]. Mol Clin Oncol, 2015, 3: 793–796. DOI:10.3892/mco.2015.536 |
| [22] | Rui M, Jiang C, Liang Y, et al. Sorafenib:a potential therapeutic drug for hepatic fibrosis and its outcomes[J]. Biomed Pharmacother, 2017, 88: 459–468. DOI:10.1016/j.biopha.2017.01.107 |
| [23] | Neef M, Ledermann M, Saegesser H, et al. Oral imatinib treatment reduces early fibrogenesis but does not prevent progression in the long term[J]. J Hepatol, 2006, 44: 167–175. DOI:10.1016/j.jhep.2005.06.015 |
| [24] | Zhao CQ. Effect and Mechanism of Secretory Fibroblast Growth Factor 21 and 19/15 in Alcoholic Livers Diseases (分泌型成纤维细胞生长因子21和19/15在酒精性肝病中的作用调节机制)[D]. Changchun: JiLin University, 2015. |
| [25] | Sanyal A, Charles ED, Neuschwander-Tetri B, et al. LBO-02-BMS-986036(pegylated FGF21) in patients with nonalcoholic steatohepatitis:a phase 2 study[J]. J Hepatol, 2017, 66: S89–S90. |
| [26] | Liu Y, Wu J, Li Z, et al. Tolerability and pharmacokinetics of hydronidone, an antifibrotic agent for hepatic fibrosis, after single and multiple doses in healthy subjects:an open-label, randomized, dose-escalating, first-in-human study[J]. Eur J Drug Metab Pharmacokinet, 2017, 42: 37–48. DOI:10.1007/s13318-015-0316-z |
| [27] | Cao W, Yan L, Wang W, et al. Simvastatin inhibits activation of hepatic stellate cells and promotes activation of adenosine monophosphate-activated protein kinase[J]. Chin J Hepatol (中华肝脏病杂志), 2012, 20: 304–309. |
| [28] | Li CY, Chen CP, Shen CY. Dynamic expression of cathepsin B in hepatic stellate cells (HSC-T6) and its significance[J]. J Med Res (医学研究杂志), 2012, 3: 36–41. |
| [29] | Cathepsin B Inhibitor: VBY-376[EB/OL]. Virobay Company official website: http://www.virobayinc.com/VBY-376.php. |
| [30] | Friedman S, Sanyal A, Goodman Z, et al. Efficacy and safety study of cenicriviroc for the treatment of non-alcoholic steatohepatitis in adult subjects with liver fibrosis:CENTAUR phase 2b study design[J]. Contemp Clin Trials, 2016, 47: 356–365. DOI:10.1016/j.cct.2016.02.012 |
| [31] | Cannito S, Novo E, Parola M. Therapeutic pro-fibrogenic signaling pathways in fibroblasts[J]. Adv Drug Deliv Rev, 2017, 121: 57–84. DOI:10.1016/j.addr.2017.05.017 |
| [32] | Neuschwander-Tetri BA, Rohit L, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, nonalcoholic steatohepatitis (FLINT):a multicentre, randomised, placebo-controlled trial[J]. Lancet, 2015, 385: 956–965. DOI:10.1016/S0140-6736(14)61933-4 |
| [33] | Loomba R, Kayali Z, Noureddin M, et al. Acetyl-CoA Carboxylase Inhibitor GS-0976 Leads to Significant Improvements in MRI-PDFF in a Phase 2, Randomized, PlaceboControlled Trial of Patients with NASH[R]. Washington: AASLD, 2017. |
| [34] | Armstrong MJ, Gaunt P, Aithal GP, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN):a multicentre, double-blind, randomised, placebocontrolled phase 2 study[J]. Lancet, 2016, 387: 679–690. DOI:10.1016/S0140-6736(15)00803-X |
| [35] | Lefebvre E, Gottwald M, Lasseter K, et al. Pharmacokinetics, safety, and CCR2/CCR5 antagonist activity of cenicriviroc in participants with mild or moderate hepatic impairment[J]. Clin Transl Sci, 2016, 9: 139–148. DOI:10.1111/cts.2016.9.issue-3 |
| [36] | Barreyro FJ, Holod S, Finocchietto PV, et al. The pancaspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis[J]. Liver Int, 2015, 35: 953–966. DOI:10.1111/liv.2015.35.issue-3 |
| [37] | Frenette C, Morelli G, Shiffman M, et al. Emricasan (IDN-6556) orally for three months in patients with cirrhosis and meld scores 11-18 improves clinical parameters of cirrhosis in patients with baseline meld score ≥ 15[J]. J Hepatol, 2016, 64: S210. |
| [38] | Zoubek ME, Trautwein C, Strnad P. Reversal of liver fibrosis:from fiction to reality[J]. Best Pract Res Clin Gastroenterol, 2017, 31: 129–141. |
| [39] | Loomba R, Lawitz E, Mantry PS, et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis:a randomized, phase 2 trial[J]. Hepatology, 2017. DOI:10.1002/hep.29514 |
| [40] | Ratziu V, Harrison SA, Francque S, et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening[J]. Gastroenterology, 2016, 150: 1147–1159. DOI:10.1053/j.gastro.2016.01.038 |
| [41] | Duan XY, Fan JG. Advances in research on relationship between galectin-3 and liver diseases[J]. World Chin J Dig (世界华人消化杂志), 2009, 17: 1909–1912. |
| [42] | Harrison SA, Marri SR, Chalasani N, et al. Randomised clinical study:GR-MD-02, a galectin-3 inhibitor, vs placebo in patients having non-alcoholic steatohepatitis with advanced fibrosis[J]. Aliment Pharm Ther, 2016, 44: 1183–1198. DOI:10.1111/apt.2016.44.issue-11-12 |
| [43] | Jiang JX, Chen X, Serizawa N, et al. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831 a novel NOX4/NOX1 inhibitor in vivo[J]. Free Radic Biol Med, 2012, 53: 289–296. DOI:10.1016/j.freeradbiomed.2012.05.007 |
| [44] | Colmenero J, Bataller R, Sanchobru P, et al. Effects of losartan on hepatic expression of nonphagocytic NADPH oxidase and fibrogenic genes in patients with chronic hepatitis C[J]. Am J Physiol-Gastr L, 2009, 297: G726–G734. |
| [45] | Chen XY, Yang CQ. Progress in the pathogenesis of liver fibrosis[J]. Chin J Clin Hepatol (临床肝胆病杂志), 2016, 19: 121–124. |
| [46] | Xu MH, Cao W. Effect of halofuginone on the change of mRNA of collagen I, tissue inhibitor of metalloproteinase-1 and α-smooth muscle actin in cirrhosis rats[J]. Suzhou Univ J Med Sci (苏州大学学报), 2009, 29: 256–258. |
| [47] | Ikenaga N, Peng ZW, Vaid KA, et al. Original article:selective targeting of lysyl oxidase-like 2(LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal[J]. GUT, 2017, 66: 1697–1708. DOI:10.1136/gutjnl-2016-312473 |
| [48] | Meissner EG, Mclaughlin M, Matthews L, et al. Simtuzumab treatment of advanced liver fibrosis in HIV and HCV-infected adults:results of a 6-month open-label safety trial[J]. Liver Int, 2016, 36: 1783–1792. DOI:10.1111/liv.13177 |
| [49] | Jouihan H, Will S, Guionaud S, et al. Superior reductions in hepatic steatosis and fibrosis with co-administration of a glucagon-like peptide-1 receptor agonist and obeticholic acid in mice[J]. Mol Metab, 2017, 6: 1360–1370. DOI:10.1016/j.molmet.2017.09.001 |
| [50] | Cao W, Li Y, Li M, et al. Txn1, Ctsd and Cdk4 are key proteins of combination therapy with taurine, epigallocatechin gallate and genistein against liver fibrosis in rats[J]. Biomed Pharmacother, 2017, 85: 611–619. DOI:10.1016/j.biopha.2016.11.071 |
| [51] | Cao W, Liao M, Zhou Y, et al. Therapeutic effects and mechanisms of combined therapy of taurine and genistein on liver fibrosis in rats[J]. Chin J Exp Tradit Med Formul (中国验方学学报), 2015, 21: 107–111. |