 2018, Vol. 53
2018, Vol. 53



药物代谢研究在创新药物研发中占据重要作用。药物进入体内后, 在体内多种代谢酶的作用下代谢生成极性更大的化合物, 促进药物排泄[1]。在药物代谢中, 细胞色素P450酶系是最重要的酶家族, 参与约75%临床药物的Ⅰ相代谢; 其次是酯酶, 参与约10%的临床药物代谢, 这些药物结构中通常含有酯键、酰胺键或硫酯键[2]。一方面, 随着人们对药物代谢认识的增加, 在新药研发中, 倾向于选择不会被P450酶代谢的化合物, 避免P450代谢酶遗传多态性以及药物相互作用的潜在影响, 提高化合物的成药性。另一方面, 根据活性物质设计合适的前药也成为创新药物研发中的一大策略, 如吉利德公司设计合成的替诺福韦的前药替诺福韦艾拉酚胺(TAF)[3]和江苏威凯尔医药科技有限公司研发的维卡格雷[4]。随着创新药物中含有酯键、酰胺键的化合物比例越来越高, 对酯酶的深入研究不仅具有科学意义, 而且是新药研发中迫切需要解决的关键问题。不同类型的酯酶在药物代谢中的作用已有研究, 人体血浆中主要的酯酶是丁酰胆碱酯酶和对氧磷酶, 水解阿司匹林和班布特罗等药物, 已有文章对其进行了综述[5]。本文主要关注肝脏和肠道酯酶在药物代谢及新药研发中的作用, 总结其种类和分布、肝脏和肠道酯酶对药物的水解作用、种属差异、酯酶活性的个体差异等。
1 酯酶的分类和组织分布 1.1 羧酸酯酶1哺乳类动物体内的酯酶是一个多基因家族, 在体内广泛分布。羧酸酯酶(carboxyles terase, CES, EC 3.1.1.1)是其中一类重要的丝氨酸水解酶家族, 主要分布于多种组织细胞的内质网肠腔侧, 是一种膜蛋白, 不分泌到血浆中。CES由多种同工酶组成, 其亚型分为5个家族: CES1、CES2、CES3、CES4A和CES5A[6]。CES1相对分子质量约60 kDa, 以180 kDa的三聚体形式存在。CES1主要表达在肝脏和肺, 肠道几乎无表达[7]。CES1一方面介导内源性物质的代谢, 如介导胆固醇酯和甘油三酯的水解[8], 进而在脂质代谢平衡中发挥重要作用, 是酯类代谢调控、肥胖和糖尿病治疗的关键靶点。而CES1的功能紊乱与动脉粥样硬化和高胆固醇血症有关[8]。另一方面, CES1在代谢外源性物质方面发挥了重要作用, 可以代谢多种药物, 如咪达普利[9]、奥司他韦[10]、伊立替康[11]等, 也与酯类药物的个性化安全用药息息相关。
1.2 羧酸酯酶2除了CES1, CES2也在药物代谢中起到了重要的作用。CES2的相对分子质量为60 kDa。其组织分布与CES1不同, CES2主要分布于小肠, 约占肠道代谢酶总量的1/3, 其次是表达在肾脏, 但在肝脏的表达量有限[7]。作为肠道主要的羧酸酯酶亚型, CES2介导大部分口服前药进入血液循环前的首过水解代谢过程, 尤其是抗肿瘤前药, 如吉西他滨的前药LY2334737[12]。CES2在药物经胃肠道上皮细胞吸收过程的代谢活化中发挥重要作用。
1.3 芳基乙酰胺脱乙酰酶人体芳基乙酰胺脱乙酰酶(arylacetamide deacetylase, AADAC)是一种微粒体丝氨酸酯酶, 相对分子质量为45 kDa, 主要表达部位为肝脏和肠道[13], 其次是膀胱[14]。与CES不同, AADAC是一种Ⅱ型膜蛋白, N-端含有未清除的信号锚定序列, 从而保留在内质网的腔体侧[15]。AADAC酶的催化活性口袋与激素敏感性脂解酶(hormone-sensitive lipase)有高度同源性, 因此, AADAC被认为是一种脂肪酶。Tiwari等[16]证实, 当表达在酵母菌时, AADAC可以水解胆固醇酯类。日本科学家Fukami团队近年来研究发现AADAC也参与几种临床药物如氟他胺[13]、非那西丁[17]、利福平[18]和酮康唑[19]的水解, 其水解代谢物被认为与其肝毒性或肾毒性相关。AADAC水解利福平生成人体内主要代谢物25-去乙酰基利福平是解毒途径, 因为与原形相比, 25-去乙酰基利福平几乎无细胞毒性[18]。据报道, 25-去乙酰基利福平是人体血浆中的主要代谢物, 但是在大鼠和兔血浆几乎检测不到[20], 因此25-去乙酰利福平可能是由人AADAC代谢生成的人体内特有的代谢物。在进行药物水解酶研究时, 需要关注种属差异性。与其他酯酶相比, 目前报道的AADAC研究结果尚少。
2 羧酸酯酶和芳基乙酰胺脱乙酰酶对药物的水解作用在人体肝脏和小肠中, 除了酯酶, 多种与药物代谢相关的酶也有表达, 如细胞色素P450酶、UDP-葡萄糖醛酸转移酶(UGT)、硫酸转移酶, 以及一些与药物转运相关的转运体, 如P-gp、MRP2和BCRP。肝脏和小肠的酯酶与Ⅱ相代谢酶和药物转运体共同参与了药物在体内的代谢、转运和排泄过程。与UGT酶相似, CES和AADAC的表达部位在内质网膜的腔体侧[21]。抗肿瘤药CPT-11经CES水解后生成的活性代谢物SN-38[11], 同时又是UGT1A1的底物[22], 进而SN-38生成之后就直接在内质网膜的腔体侧经UGT1A1催化生成葡萄糖醛酸结合物。除此之外, 经CES水解生成的有机阴离子可能是MRP2或BCRP的底物, 进而被排出体外。对于酶-酶相互作用或酶-转运体相互作用来说, CES是一种重要的药物代谢相关酶。
水解反应通常是酯类药物在体内处置过程的第一步, 水解产物可能具有药理活性或不良反应, 因此鉴定参与酯类药物水解的酯酶表型对了解药物的代谢特征非常重要。临床前多种动物的药动学和药效学数据是新药(特别是前药)进行人体试验的重要参考数据, 因此阐明相关酯酶亚型的生物化学性质是非常重要的。另外, 通过总结酯类药物的水解酶表型和酯酶的底物特异性有助于设计更理想的前体药物。
2.1 底物特异性4-硝基苯基乙酸酯(PNPA)和7-乙酰氧基-4-甲基香豆素(4-MUA)是常用的非选择性酯酶底物[23, 24], 常作为工具药使用, 一般用于考察抑制剂对总体酯酶的抑制作用。尽管CES1和CES2活性催化位点的氨基酸同源性很高, 但两者的底物选择性却不同。另一方面, 底物特异性受酰基-酶结合体容量的限制, 在酯酶的活性口袋存在构象干扰, 底物特异性区别不是很严格[21]。一般来说, CES1倾向于水解含有大酰基小醇基的底物药物, 如氯吡格雷[25]、非诺贝特[26]、奥昔布宁[27]和奥司他韦[10]等。CES2底物药物特点与CES1相反, 多含有小酰基大醇基结构, 如CPT-11[11]、普拉格雷[28]和普鲁卡因[26, 29]等。AADAC的底物一般为小酰基大醇基结构, 与CES2的底物性质相似[26]。AADAC与CES2的某些底物存在重叠, 如AADAC和CES2可以共同参与氟他胺[13]和普拉格雷[30]的水解, 但是非那西丁[17]、利福霉素[18]和茚地普隆[31]仅由AADAC水解。
对于一个特定的药物结构, 酯酶的选择性与酯键结构的空间位阻有关。例如维卡格雷, 其结构中两个酯键分别由CES1和CES2催化代谢。维卡格雷结构中苄位的酯键与氯吡格雷相同, 由肝脏的CES1特异性代谢[32]; 而噻吩环上的酯键却表现出CES2代谢的特异性[4], 经CES2代谢的清除率是CES1的10倍以上[4]。维卡格雷在肠道吸收过程中经历完全的首过代谢[33], 因此CES2在水解维卡格雷生成2-氧代氯吡格雷途径中发挥了重要作用。同时有多个酯酶参与一个药物的水解代谢是很常见的, 因此需要在药物研发过程中鉴定参与药物水解的酯酶, 不仅帮助预测药物-药物相互作用(DDI), 同时对药物的设计提供思路。表 1[9, 11, 13, 17, 18, 26-29, 31, 32, 34]列出了部分目前已报道的CES1、CES2和AADAC的典型底物及其水解动力学参数等信息。
| Table 1 Kinetic parameters of probe substrates for human carboxylesterase (CES) and arylacetamide deacetylase (AADAC) |
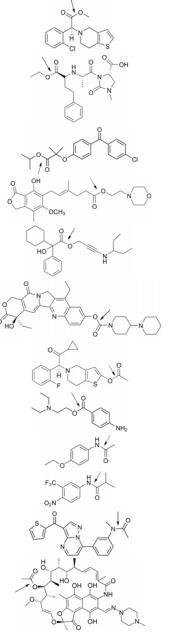
根据酯酶的底物特异性规则, 从化合物的结构可以大概推测其酯酶表型。但是该规则并不是完全适用于所有化合物, 如抗孕药双炔失碳酯, 其结构中的酯键类型是大的醇基和小的丙酰基, 符合CES2的底物特征, 但实际上肝脏中CES1的贡献率约90.7%[35], 远高于CES2。因此, 确定一个化合物的酯酶表型时, 除了利用底物特异性规则来大概推测参与代谢的酯酶类型, 还需要通过具体的实验来验证。
多个酯酶可能同时参与药物的水解代谢过程, 因此特异性抑制剂一般作为工具药来考察药物的酯酶表型, 并评价各个酶的贡献。考察药物的水解酶表型时, 一般从两个方面来验证, 首先考察药物在各个重组酶体系中的水解速率, 再在人肝微粒体或肠微粒体的孵化体系中加入各个酯酶的特异性抑制剂, 从另一方面验证药物的酶表型。目前发现的唯一一个CES1特异性抑制剂是洋地黄皂苷, 在重组CES1中抑制利多卡因的IC50为25.8 μmol·L-1[36], 抑制PNPA的IC50为9.2 μmol·L-1[36]。但是在人肝微粒体的孵化体系中却不能抑制CES1的特异性底物利多卡因和非诺贝特的水解, 可能由于其他蛋白的干扰表现不出抑制活性[36]。CES2的特异性抑制剂有黄酮哌酯、洛哌丁胺、替米沙坦[36]和非诺贝特[37], 但黄酮哌酯在酶体系中也会水解, 水解产物对AADAC和CES都有一定的抑制作用, 因此黄酮哌酯并不是一个很好的特异性CES2抑制剂。替米沙坦在人肝微粒体(HLM)中抑制CPT-11水解的IC50约0.5 μmol·L-1, 在重组CES2中IC50约0.4 μmol·L-1, 是一个专属性的强抑制剂[36]。洛哌丁胺抑制CES2的IC50约0.38 μmol·L-1[38]。长春碱和毒扁豆碱经常作为AADAC的特异性抑制剂使用[36, 39], 但两者对CES2酶也有一定的抑制作用[36], 目前为止, 还未发现AADAC的特异性抑制剂。BNPP (双-p-硝基苯基磷酸盐)和DDVP [O, O-二甲基-O-(2, 2-二氯乙烯基)磷酸酯]不仅是羧酸酯酶的广泛抑制剂, 还可以抑制血浆中水解酶的活性, 如对氧磷酶和丁酰胆碱酯酶, 常用的浓度为0.1~1 mmol·L-1。二异丙基氟磷酸(DFP)在100 μmol·L-1浓度时可以抑制CES和AADAC的活性[40]。苯甲基磺酰氟(PMSF)在100 μmol·L-1浓度时可以抑制CES活性[34], 但是不抑制AADAC活性[13]。降脂类药物辛伐他汀和美伐他汀可抑制CES及AADAC活性[41, 42]。根据不同的化学抑制剂对酯酶的选择性抑制, 可以在一个混合的酶体系(如人肝微粒体或肠微粒体)加入单个或多个抑制剂, 以评价各个酶的贡献。表 2总结了部分抑制剂的作用酶及抑制的IC50值。
| Table 2 Chemical inhibitors for CES or AADAC and IC50 values. BNPP: bis-p-Nitrophenyl phosphate; PNPA: p-Nitrophenyl acetate; CPT-11: Irinotecan |
哺乳动物肝脏的酯酶对酯类化合物的水解起到了重要的作用, 也参与内源性的短链或长链酰基甘油、长链肉毒碱、长链乙酰辅酶A酯等物质的水解[43-45]。人体肝脏中同时表达有CES1、CES2和AADAC。CES1含有一个灵活的和刚性的口袋, 很多化合物可以结合CES1。被CES1催化的同时可能会发生转酯现象, 特别是对于疏水性醇, 在底物被CES1水解的同时即发生了转酯反应[7]。Brzezinski等[46]研究发现, 在乙醇存在时, CES1可以催化可卡因的转酯反应, 生成活性代谢物可卡乙碱。在水解可卡因的两步反应中, 可卡因的羧基甲酯首先与CES1生成了共价的酰基-酶中间体, 然后乙醇的羟基进攻该中间体, 与羰基结合重新生成一个酯结构的代谢物, 即可卡乙碱[47]。在类似的情况下, 一些化合物在被水解酶代谢的同时可能会产生一个新的酯类化合物。相比CES1, CES2几乎无转酯活性。近几年受到专注的一种肝脏酯酶是芳基乙酰胺脱乙酰酶(AADAC)。C57BL/6J小鼠体内高铁血红蛋白水平与非那西丁的水解代谢物对氨基苯乙醚浓度水平相关[39]; 提前给予水解酶的抑制剂后, 小鼠血浆中高铁血红蛋白和对氨基苯乙醚的生成均下降[39]。推测非那西丁经AADAC水解后继续由CYP1A2和CYP2E1催化生成羟胺结构, 进而产生毒性反应[39]。
前药一般被设计用来提高生物利用度或改善药动学特性。霉酚酸酯[34]、吉西他滨的前药LY2334737[12]等在肠道或肝脏被CES部分水解, 从而释放出活性成分。人体肠道含有的酯酶类型为CES2和AADAC[21, 30], 若一个口服药物是CES1的底物, 则通过胃肠道吸收时不会被水解代谢, 而在肝脏代谢, 如替莫普利[48]; 而若一个口服药物是CES2或AADAC的底物, 由于肠道的水解能力较弱, 一般经过肠道吸收时会被部分水解代谢, 剩余的部分在肝脏被代谢, 如AADAC的底物氟他胺[13]。人体肝脏中同时表达有CES1、CES2和AADAC, 其蛋白表达水平表现出很大的个体差异。由于底物选择性和蛋白表达量的差异, 各个酯酶亚型对药物水解的贡献率是不同的。Watanabe和Kurokawa等[17, 30]提供了计算各个酯酶对药物水解过程的贡献率的方法。该方法称为相对活性因子法(RAF), 首先需要测得待测物在各个混合基质(人肝微粒体或肠微粒体)和纯化的重组酶的水解速率, 而各个酶的RAF值为探针底物(部分探针底物总结在表 1中)在混合基质和纯化的重组酶中的水解速率比值, 将RAF值作为校正因子参与待测物贡献率的计算, 从而计算得到各个酶对待测物水解过程的贡献率[30]。如Kurokawa等[30]选取CES1、CES2和AADAC的探针底物为非诺贝特、普鲁卡因和茚地普隆, 计算的RAF值分别为0.83、0和2.60 [人肠微粒体(HIM)与重组酶之间比较], 最终得到普拉格雷在HIM中水解时AADAC和CES2的贡献率分别为50.6%和46.4%。一些研究者认为, 不能单纯的以水解速率来考察酯酶的贡献活性, 应该考虑各个酯酶的实际表达量。但是目前报道的肝脏酯酶蛋白表达量数据差异较大[49, 50], 如人肝微粒体中CES1的平均表达量有报道约为10 pmol·mg-1蛋白[50], 也有报道约为360 pmol·mg-1蛋白[49], 报道的表达量水平差异30倍以上, 无法达成统一的基准, 且AADAC在肝脏的表达量未见报道, 因此推荐使用RAF法计算酯酶的贡献率。
2.4 肠道酯酶在药物吸收过程中作用对于前药和易水解药物的口服给药过程来说, 肠道的酯酶对其首过代谢产生了重要的影响。肠道上皮细胞表达有CES2和AADAC酶[21, 30], 对于易水解药物, 如普拉格雷, 在肠道吸收过程中经历了完全的首过代谢, 生成水解代谢物普拉格雷硫代内酯并且被进一步氧化代谢[30]。而对于相对难水解的前药, 如氟他胺, 肠道吸收时仅经历部分首过代谢[13], 未被水解部分进入体循环[51]。氯吡格雷和普拉格雷虽然结构相似, 却表现出了不同的吸收特征。氯吡格雷结构中苄位的酯基被CES1选择性代谢[32], 不会被肠道的CES2或AADAC水解代谢, 因此经肠道吸收时未经历水解代谢过程, 而是在肝脏被水解生成羧酸代谢物[32]。普拉格雷的酯基在噻吩环上, 易被CES2或AADAC水解[30], 因此在肠道吸收时经历完全的首过代谢, 人体血浆中几乎检测不到普拉格雷[28]。由此可以看出, 结构相似化合物的代谢途径并不是一致的, 利用酯酶的底物特异性和组织分布的特点, 合理地设计药物可以加速新药的研发。
对介导药物水解代谢的酯酶研究是一个逐渐深入的过程。对普拉格雷而言, Williams等[28]证实其水解代谢酶为CES1和CES2, 并且利用纯化的重组酶考察了水解动力学特征。普拉格雷在穿过体外Caco-2单层细胞的顶膜侧到达基底侧时, 完全转化为水解代谢物R-95913。而在后续的研究中发现普拉格雷在比格犬的肠道中可以水解, 而犬的肠道不含有CES活性[52], 由此推测肠道还有除CES2外的其他酶参与普拉格雷的水解代谢。Kurokawa等[30]发现人肠AADAC酶也参与普拉格雷的水解过程, 利用RAF法计算得到其贡献率为50%左右, CES2的贡献率为46%左右。该结果也说明, 虽然比格犬的肠道不含有CES活性, 但是含有相当量的AADAC活性, 因此在选择物种进行临床前药动学评价时需要引起关注。Kazui等[53]发现除了已被报道的酯酶参与普拉格雷水解代谢, 人体肠道的Raf激酶抑制蛋白(RKIP)也参与其水解过程, 并将该酶从人小肠S9中分离出来, 利用LC-MS/MS确证了RKIP的氨基酸序列[53]。越来越多的参与药物代谢的酯酶将会被发现, 有助于药物代谢研究和安全性评价。
考察药物在人体肠道的吸收过程时, 通常使用Caco-2细胞模型。Caco-2细胞系是一个快速筛选药物在肠道吸收特征的体外模型[54]。可以得到表观渗透系数(Papp)和表观渗透系数的比率(Papp ratio = Papp(B→A)/Papp(A→B)), 用以评价药物在人肠道细胞中的渗透性和是否有肠道外排转运体的参与。该模型在协助药物开发阶段发挥了很大的作用, 但是对于酯类药物而言, Caco-2细胞模型不能考察药物的吸收过程, 因为在Caco-2细胞系中, 虽然CES1和CES2的mRNA和蛋白表达水平都呈现时间依赖性的趋势, 但是CES1的表达水平总是高于CES2[55]。然而在人体肠道细胞中是相反的情况, 肠道上皮细胞几乎不表达CES1, 而高表达有CES2和AADAC[21, 30]。替莫普利在穿过Caco-2细胞时会很快水解为替莫普利拉, 因为其是CES1的典型底物, 但其实际上在人体小肠内不水解[48]。因此, 在Caco-2细胞系中评价一个CES1底物的细胞渗透性是非常不准确的。考察药物在肠道的吸收特征对于新药的药性评价非常重要, 但是需要关注体外模型和实验动物的选择。
3 种属差异在各种属肝脏CES比较中, 猴CES1、比格犬CES D1和兔酯酶1与人肝脏CES1的同源性约92.9%、79.7%和81.1%[52]。大鼠的血浆和组织中都含有丰富的羧酸酯酶, 大鼠CES1家族的水解酶A、B、C和egasyn与人CES1有77.6%、67.0%、67.4%和75.0%的同源性[52]。值得注意的是, 比格犬小肠中无CES活性[52], 但是有AADAC活性[30], 因此选用比格犬做实验动物时, 其表现出的药物动力学特征可能与其他种属有差异。就血浆中酯酶种类而言, 小鼠、大鼠、兔、马等动物的血浆中都含有丰富的CES活性, 但是人血浆中不含CES[5]。人血浆中含有的酯酶类型与比格犬血浆相似, 主要为丁酰胆碱酯酶(BChE)、对氧磷酶(PON1)和白蛋白[5]。
由于酯酶在各种属体内的组织分布存在差异, 因此对于一个特定的药物来说可以产生药动学差异。如比格犬体内维卡格雷活性代谢物的暴露量是大鼠的6倍以上, 苄位和噻吩环上酯键水解后的羧酸代谢物(CAM)的暴露量是大鼠的25%左右[56]。产生这一现象的原因是维卡格雷在比格犬肠道水解为2-氧-氯吡格雷后不再继续水解为CAM, 而是在肠道CYP450的作用下生成活性代谢物。大鼠的肠道和血浆中都分布有丰富的羧酸酯酶, 因此在维卡格雷水解为2-氧-氯吡格雷后会继续水解为CAM, 使活性代谢物的生成量减少。卡培他滨口服后在肠道和肝脏都会水解为5'-DFCR, Shindoh等[57]发现在人和猴肠道的水解清除率比肝脏低6倍左右, 而大鼠和小鼠体内则表现出相反的现象, 肠道的清除率高于肝脏。由于大鼠和小鼠的肝脏和肠道中胞苷脱氨酶活性很低, 而猴与人对卡培他滨的水解表现相似的特点, 因此选择猴作为模型动物来评价卡培他滨的安全性是比较合适的。Liu等[58]发现治疗风湿性关节炎的候选药物GDC-0834的酰胺水解代谢物M1在临床前实验动物体内浓度很低, 大鼠、比格犬、猴体内M1与原形暴露量比值约1.5%、26%、0。但是在人体内M1却是主要的药物相关物质, 体内暴露量很高, 且原形药物血浆中浓度低于定量下限(< 1 ng·mL-1)[58], 酰胺水解是GDC-0834在人体内主要代谢途径。体外考察测得人肝微粒体中M1的生成速率比大鼠、比格犬和猴高23~169倍[58]。GDC-0834的例子说明虽然临床前考察的几种动物模型都显示出相似的代谢和药动学特征, 但是与人体的代谢差异很大。不同种属体内酯酶的组织分布和种类不同, 因此选择种属考察药物(特别是前药)体内ADME时需谨慎。
4 酯酶活性的个体差异健康人体中CES1在肝组织中高分布, 血浆中仅有少量分布; 但肝细胞受损后血中CES1会急剧升高, 因此CES1可作为急性肝损伤及肝癌等肝脏疾病早期诊断的标志物。我国的一些研究者也根据CES1和CES2的特点合成了专属性荧光探针, 可专一、实时地反映特定环境下目标酶的活性, 从而实现对特定功能蛋白实时定量监测的目的[59-61]。
由于羧酸酯酶表达量的个体差异和基因多态性等原因, 导致酯酶活性存在较大的个体差异。Hines等[50]发现CES1在白种人、非裔美国人和西班牙人肝微粒体中的表达量分别为17.95、14.99和10.86 pmol·mg-1蛋白, 在白种人肝脏中表达高于其他两个人种[50]; 而CES2在3个人种的表达分别为2.99、2.79和1.79 pmol·mg-1蛋白, 表达量比值CES1/CES2约6左右[50]。近期一些研究利用液相色谱联合质谱检测(LC-MS/MS)的方法可准确地测得肝脏内CES的表达量。Sato等[62]利用LC-MS/MS法测得CES1和CES2在人肝微粒体中的平均表达量为363和22.2 pmol·mg-1蛋白, 表达量比值CES1/CES2约16左右; Wang等[49]同样利用LC-MS/MS法, 测得CES1在肝组织中平均表达量约176 pmol·mg-1蛋白, 与Hines等测得的数据(蛋白印迹法)相差很大。CES在不同个体间的表达差异很大, CES1表达量的差异在5~10倍左右[50, 62, 63], CES2表达量差异在3~6倍左右[62, 64]。尽管不同研究者测得数据差异明显, 但是从中可以看出, CES1和CES2在不同个体间的表达水平差异显著, 且CES1的表达量是CES2的10倍左右。而AADAC在肝脏中的蛋白表达量没有报道。
除了个体间蛋白表达量差异外, 酯酶的活性与基因多态性相关。Zhu等[65]发现CES1的两个突变体p.Gly143Glu和p.Asp260fs对其活性有显著影响, 完全丧失对哌甲酯的水解能力, 对PNPA的水解活性降低至野生型的21.4%和0.6%。而突变体p.Gly143Glu在白种人、黑种人、西班牙人和亚洲人种的出现频率分别为3.7%、4.3%、2.0%和0[64]。在健康白种人志愿者中, 突变体p.Gly143Glu携带者服用氯吡格雷后, 羧酸代谢物CAM的暴露量为不携带突变基因的53%, 活性代谢物的暴露量升高67%[66], 表明在携带突变基因的人中, 氯吡格雷的水解代谢被抑制, 从而提高了活性代谢物的暴露量。CES2的R34W和V142M氨基酸的替换可能导致水解CPT-11的能力丧失[67]。Kubo等[67]研究发现, 体外野生型CES2重组酶水解CPT-11的清除率约92 pmol·min-1·nmol-1, 而R34W和V142M突变CES2酶的清除率小于5.0 pmol·min-1·nmol-1。羧酸酯酶基因多态性的存在可能会造成显著的临床上药动学和药物响应的改变。
5 结语与展望为避免由CYP450酶代谢引起的药物-药物相互作用以及基因多态性造成的药动学变化, 新药研发中设计出了越来越多的酯类化合物。酯水解过程对不同的化合物可能是解毒、活化或致毒过程, 与药物的有效性和安全性密切相关。人体肝脏和肠道中主要的酯酶是羧酸酯酶(CES)和芳基乙酰胺脱乙酰酶(AADAC)。对AADAC酶的研究起步较晚, 但是从近年来发表的研究成果来看, AADAC对药物的吸收、代谢过程起到的作用不容忽视。不同酯酶有较明显的底物特异性, 此规律可以指导新药的合理设计, 或者推测各个酶对化合物水解过程的贡献。目前观察到CES和AADAC存在广泛的种属差异, 对临床前药物的安全性和药动学评价, 需慎重选择动物模型。在个体差异方面, 酯酶的蛋白表达量和基因多态性严重影响其水解活性, 携带突变基因的患者将成为某些酯酶底物的弱代谢者。这些因素使得预测个体患者的药动学特征和药物响应变得困难。不仅如此, 几乎没有酯类药物单独设计临床试验以评价基于酯酶的药物-药物相互作用, 这可能会低估DDI产生的影响。
本文通过具体的实例展示了近年来国内外学者对酯酶的研究进展, 以期为含酯键药物的合理设计提供理论指导, 加速新药研发进程。
| [1] | Guengerich FP. Cytochrome P450s and other enzymes in drug metabolism and toxicity[J]. AAPS J, 2006, 8: E101–E111. DOI:10.1208/aapsj080112 |
| [2] | Williams JA, Hyland R, Jones BC, et al. Drug-drug interactions for UDP-glucuronosyltransferase substrates:a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios[J]. Drug Metab Dispos, 2004, 32: 1201–1208. DOI:10.1124/dmd.104.000794 |
| [3] | Birkus G, Kutty N, He GX, et al. Activation of 9-[(R)-2-[[(S)-[[(S)-1-(isopropoxycarbonyl)ethyl]amino]phenoxyphosphinyl]-methoxy]propyl]adenine (GS-7340) and other tenofovir phosphonoamidate prodrugs by human proteases[J]. Mol Pharmacol, 2008, 74: 92–100. DOI:10.1124/mol.108.045526 |
| [4] | Qiu ZX, Li N, Song L, et al. Contributions of intestine and plasma to the presystemic bioconversion of vicagrel, an acetate of clopidogrel[J]. Pharm Res, 2014, 31: 238–251. DOI:10.1007/s11095-013-1158-5 |
| [5] | Li B, Sedlacek M, Manoharan I, et al. Butyrylcholinesterase, paraoxonase, and albumin esterase, but not carboxylesterase, are present in human plasma[J]. Biochem Pharmacol, 2005, 70: 1673–1684. DOI:10.1016/j.bcp.2005.09.002 |
| [6] | Holmes RS, Wright MW, Laulederkind SJ, et al. Recommended nomenclature for five mammalian carboxylesterase gene families:human, mouse, and rat genes and proteins[J]. Mamm Genome, 2010, 21: 427–441. DOI:10.1007/s00335-010-9284-4 |
| [7] | Imai T. Human carboxylesterase isozymes:catalytic properties and rational drug design[J]. Drug Metab Pharmacokinet, 2006, 21: 173–185. DOI:10.2133/dmpk.21.173 |
| [8] | Satoh T, Hosokawa M. Carboxylesterases:structure, function and polymorphism in mammals[J]. J Pestic Sci, 2010, 35: 218–228. DOI:10.1584/jpestics.R10-02 |
| [9] | Takahashi S, Katoh M, Saitoh T, et al. Allosteric kinetics of human carboxylesterase 1:species differences and interindividual variability[J]. J Pharm Sci, 2008, 97: 5434–5445. DOI:10.1002/jps.21376 |
| [10] | Zhu H, Markowitz JS. Carboxylesterase 1(CES1) genetic polymorphisms and oseltamivir activation[J]. Eur J Clin Pharmacol, 2013, 69: 733–734. DOI:10.1007/s00228-012-1350-2 |
| [11] | Humerickhouse R, Lohrbach K, Li L, et al. Characterization of CPT-11 hydrolysis by human liver carboxylesterase isoforms hCE-1 and hCE-2[J]. Cancer Res, 2000, 60: 1189–1192. |
| [12] | Pratt SE, Durland-Busbice S, Shepard RL, et al. Human carboxylesterase-2 hydrolyzes the prodrug of gemcitabine (LY2334737) and confers prodrug sensitivity to cancer cells[J]. Clin Cancer Res, 2013, 19: 1159–1168. DOI:10.1158/1078-0432.CCR-12-1184 |
| [13] | Watanabe A, Fukami T, Nakajima M, et al. Human arylacetamide deacetylase is a principal enzyme in flutamide hydrolysis[J]. Drug Metab Dispos, 2009, 37: 1513–1520. DOI:10.1124/dmd.109.026567 |
| [14] | Kobayashi Y, Fukami T, Nakajima A, et al. Species differences in tissue distribution and enzyme activities of arylacetamide deacetylase in human, rat, and mouse[J]. Drug Metab Dispos, 2012, 40: 671–679. DOI:10.1124/dmd.111.043067 |
| [15] | Frick C, Atanasov AG, Arnold P, et al. Appropriate function of 11 beta-hydroxysteroid dehydrogenase type 1 in the endoplasmic reticulum lumen is dependent on its N-terminal region sharing similar topological determinants with 50-kDa esterase[J]. J Biol Chem, 2004, 279: 31131–31138. DOI:10.1074/jbc.M313666200 |
| [16] | Tiwari R, Koffel R, Schneiter R. An acetylation/deacetylation cycle controls the export of sterols and steroids from S. cerevisiae[J]. EMBO J, 2007, 26: 5109–5119. DOI:10.1038/sj.emboj.7601924 |
| [17] | Watanabe A, Fukami T, Takahashi S, et al. Arylacetamide deacetylase is a determinant enzyme for the difference in hydrolase activities of phenacetin and acetaminophen[J]. Drug Metab Dispos, 2010, 38: 1532–1537. DOI:10.1124/dmd.110.033720 |
| [18] | Nakajima A, Fukami T, Kobayashi Y, et al. Human arylacetamide deacetylase is responsible for deacetylation of rifamycins:rifampicin, rifabutin, and rifapentine[J]. Biochem Pharmacol, 2011, 82: 1747–1756. DOI:10.1016/j.bcp.2011.08.003 |
| [19] | Fukami T, Iida A, Konishi K, et al. Human arylacetamide deacetylase hydrolyzes ketoconazole to trigger hepatocellular toxicity[J]. Biochem Pharmacol, 2016, 116: 153–161. DOI:10.1016/j.bcp.2016.07.007 |
| [20] | Tenconi L, Beretta E. Urinary and biliary metabolites of rifampicin in different animal species[J]. Proc Eur Soc Drug Toxit, 1970, 11: 80–85. |
| [21] | Fukami T, Yokoi T. The emerging role of human esterases[J]. Drug Metab Pharmacokinet, 2012, 27: 466–477. DOI:10.2133/dmpk.DMPK-12-RV-042 |
| [22] | Jinno H, Tanaka-Kagawa T, Hanioka N, et al. Glucuronidation of 7-ethyl-10-hydroxycamptothecin (SN-38), an active metabolite of irinotecan (CPT-11), by human UGT1A1 variants, G71R, P229Q, and Y486D[J]. Drug Metab Dispos, 2003, 31: 108–113. DOI:10.1124/dmd.31.1.108 |
| [23] | Holenarsipur VK, Gaud N, Sinha J, et al. Absorption and cleavage of enalapril, a carboxyl ester prodrug, in the rat intestine:in vitro, in situ intestinal perfusion and portal vein cannulation models[J]. Biopharm Drug Dispos, 2015. DOI:10.1002/bdd.1950 |
| [24] | Nakamura Y, Sugihara K, Sone T, et al. The in vitro metabolism of a pyrethroid insecticide, permethrin, and its hydrolysis products in rats[J]. Toxicology, 2007, 235: 176–184. DOI:10.1016/j.tox.2007.03.016 |
| [25] | Farid NA, Kurihara A, Wrighton SA. Metabolism and disposition of the thienopyridine antiplatelet drugs ticlopidine, clopidogrel, and prasugrel in humans[J]. J Clin Pharmacol, 2010, 50: 126–142. DOI:10.1177/0091270009343005 |
| [26] | Fukami T, Kariya M, Kurokawa T, et al. Comparison of substrate specificity among human arylacetamide deacetylase and carboxylesterases[J]. Eur J Pharm Sci, 2015, 78: 47–53. DOI:10.1016/j.ejps.2015.07.006 |
| [27] | Sato Y, Miyashita A, Iwatsubo T, et al. Conclusive identification of the oxybutynin-hydrolyzing enzyme in human liver[J]. Drug Metab Dispos, 2012, 40: 902–906. DOI:10.1124/dmd.111.043208 |
| [28] | Williams ET, Jones KO, Ponsler GD, et al. The biotransformation of prasugrel, a new thienopyridine prodrug, by the human carboxylesterases 1 and 2[J]. Drug Metab Dispos, 2008, 36: 1227–1232. DOI:10.1124/dmd.107.020248 |
| [29] | Jewell C, Ackermann C, Payne NA, et al. Specificity of procaine and ester hydrolysis by human, minipig, and rat skin and liver[J]. Drug Metab Dispos, 2007, 35: 2015–2022. DOI:10.1124/dmd.107.015727 |
| [30] | Kurokawa T, Fukami T, Yoshida T, et al. Arylacetamide deacetylase is responsible for activation of prasugrel in human and dog[J]. Drug Metab Dispos, 2016, 44: 409–416. DOI:10.1124/dmd.115.068221 |
| [31] | Shimizu M, Fukami T, Ito Y, et al. Indiplon is hydrolyzed by arylacetamide deacetylase in human liver[J]. Drug Metab Dispos, 2014, 42: 751–758. DOI:10.1124/dmd.113.056184 |
| [32] | Zhu HJ, Wang XW, Gawronski BE, et al. Carboxylesterase 1 as a determinant of clopidogrel metabolism and activation[J]. J Pharmacol Exp Ther, 2013, 344: 665–672. DOI:10.1124/jpet.112.201640 |
| [33] | Qiu Z, Li N, Wang X, et al. Pharmacokinetics of vicagrel, a promising analog of clopidogrel, in rats and Beagle dogs[J]. J Pharm Sci, 2013, 102: 741–749. DOI:10.1002/jps.23394 |
| [34] | Fujiyama N, Miura M, Kato S, et al. Involvement of carboxylesterase 1 and 2 in the hydrolysis of mycophenolate mofetil[J]. Drug Metab Dispos, 2010, 38: 2210–2217. DOI:10.1124/dmd.110.034249 |
| [35] | Jiang J, Chen X, Zhong D. Predominant contributions of carboxylesterase 1 and 2 in hydrolysis of anordrin in humans[J]. Xenobiotica, 2017. DOI:10.1080/00498254.2017.1333658 |
| [36] | Shimizu M, Fukami T, Nakajima M, et al. Screening of specific inhibitors for human carboxylesterases or arylacetamide deacetylase[J]. Drug Metab Dispos, 2014, 42: 1103–1109. DOI:10.1124/dmd.114.056994 |
| [37] | Fukami T, Takahashi S, Nakagawa N, et al. In vitro evaluation of inhibitory effects of antidiabetic and antihyperlipidemic drugs on human carboxylesterase activities[J]. Drug Metab Dispos, 2010, 38: 2173–2178. DOI:10.1124/dmd.110.034454 |
| [38] | Quinney SK, Sanghani SP, Davis WI, et al. Hydrolysis of capecitabine to 5'-deoxy-5-fluorocytidine by human carboxylesterases and inhibition by loperamide[J]. J Pharmacol Exp Ther, 2005, 313: 1011–1016. DOI:10.1124/jpet.104.081265 |
| [39] | Kobayashi Y, Fukami T, Higuchi R, et al. Metabolic activation by human arylacetamide deacetylase, CYP2E1, and CYP1A2 causes phenacetin-induced methemoglobinemia[J]. Biochem Pharmacol, 2012, 84: 1196–1206. DOI:10.1016/j.bcp.2012.08.015 |
| [40] | Tabata T, Katoh M, Tokudome S, et al. Identification of the cytosolic carboxylesterase catalyzing the 5'-deoxy-5-fluorocytidine formation from capecitabine in human liver[J]. Drug Metab Dispos, 2004, 32: 1103–1110. DOI:10.1124/dmd.104.000554 |
| [41] | Wang XW, Zhu HJ, Markowitz JS. Carboxylesterase 1-mediated drug-drug interactions between clopidogrel and simvastatin[J]. Biol Pharm Bull, 2015, 38: 292–297. DOI:10.1248/bpb.b14-00679 |
| [42] | Fleming CD, Bencharit S, Edwards CC, et al. Structural insights into drug processing by human carboxylesterase 1:tamoxifen, mevastatin, and inhibition by benzil[J]. J Mol Biol, 2005, 352: 165–177. DOI:10.1016/j.jmb.2005.07.016 |
| [43] | Furihata T, Hosokawa M, Nakata F, et al. Purification, molecular cloning, and functional expression of inducible liver acylcarnitine hydrolase in C57BL/6 mouse, belonging to the carboxylesterase multigene family[J]. Arch Biochem Biophys, 2003, 416: 101–109. DOI:10.1016/S0003-9861(03)00286-8 |
| [44] | Hosokawa M, Suzuki K, Takahashi D, et al. Purification, molecular cloning, and functional expression of dog liver microsomal acyl-CoA hydrolase:a member of the carboxylesterase multigene family[J]. Arch Biochem Biophys, 2001, 389: 245–253. DOI:10.1006/abbi.2001.2346 |
| [45] | Mentlein R, Heymann E. Hydrolysis of ester- and amidetype drugs by the purified isoenzymes of nonspecific carboxylesterase from rat liver[J]. Biochem Pharmacol, 1984, 33: 1243–1248. DOI:10.1016/0006-2952(84)90176-X |
| [46] | Brzezinski MR, Abraham TL, Stone CL, et al. Purification and characterization of a human liver cocaine carboxylesterase that catalyzes the production of benzoylecgonine and the formation of cocaethylene from alcohol and cocaine[J]. Biochem Pharmacol, 1994, 48: 1747–1755. DOI:10.1016/0006-2952(94)90461-8 |
| [47] | Brzezinski MR, Spink BJ, Dean RA, et al. Human liver carboxylesterase hCE-1:binding specificity for cocaine, heroin, and their metabolites and analogs[J]. Drug Metab Dispos, 1997, 25: 1089–1096. |
| [48] | Imai T, Imoto M, Sakamoto H, et al. Identification of esterases expressed in Caco-2 cells and effects of their hydrolyzing activity in predicting human intestinal absorption[J]. Drug Metab Dispos, 2005, 33: 1185–1190. DOI:10.1124/dmd.105.004226 |
| [49] | Wang X, Liang Y, Liu L, et al. Targeted absolute quantitative proteomics with SILAC internal standards and unlabeled fulllength protein calibrators (TAQSI)[J]. Rapid Commun Mass Spectrom, 2016, 30: 553–561. DOI:10.1002/rcm.7482 |
| [50] | Hines RN, Simpson PM, McCarver DG. Age-dependent human hepatic carboxylesterase 1(CES1) and carboxylesterase 2(CES2) postnatal ontogeny[J]. Drug Metab Dispos, 2016, 44: 959–966. DOI:10.1124/dmd.115.068957 |
| [51] | Schulz M, Schmoldt A, Donn F, et al. The pharmacokinetics of flutamide and its major metabolites after a single oral dose and during chronic treatment[J]. Eur J Clin Pharmacol, 1988, 34: 633–636. DOI:10.1007/BF00615229 |
| [52] | Taketani M, Shii M, Ohura K, et al. Carboxylesterase in the liver and small intestine of experimental animals and human[J]. Life Sci, 2007, 81: 924–932. DOI:10.1016/j.lfs.2007.07.026 |
| [53] | Kazui M, Ogura Y, Hagihara K, et al. Human intestinal raf kinase inhibitor protein (RKIP) catalyzes prasugrel as a bioactivation hydrolase[J]. Drug Metab Dispos, 2016, 44: 115–123. |
| [54] | Artursson P, Palm K, Luthman K. Caco-2 monolayers in experimental and theoretical predictions of drug transport[J]. Adv Drug Deliv Rev, 2001, 46: 27–43. DOI:10.1016/S0169-409X(00)00128-9 |
| [55] | Ohura K, Sakamoto H, Ninomiya S, et al. Development of a novel system for estimating human intestinal absorption using Caco-2 cells in the absence of esterase activity[J]. Drug Metab Dispos, 2010, 38: 323–331. DOI:10.1124/dmd.109.029413 |
| [56] | Qiu ZX, Gao WC, Dai Y, et al. Species comparison of pre-systemic bioactivation of vicagrel, a new acetate derivative of clopidogrel[J]. Front Pharmacol, 2016, 7: 1–12. |
| [57] | Shindoh H, Nakano K, Yoshida T, et al. Comparison of in vitro metabolic conversion of capecitabine to 5-FU in rats, mice, monkeys and humans-toxicological implications[J]. J Toxicol Sci, 2011, 36: 411–422. DOI:10.2131/jts.36.411 |
| [58] | Liu L, Halladay JS, Shin Y, et al. Significant species difference in amide hydrolysis of GDC-0834, a novel potent and selective Bruton's tyrosine kinase inhibitor[J]. Drug Metab Dispos, 2011, 39: 1840–1849. DOI:10.1124/dmd.111.040840 |
| [59] | Jin Q, Feng L, Wang DD, et al. A highly selective nearinfrared fluorescent probe for carboxylesterase 2 and its bioimaging applications in living cells and animals[J]. Biosens Bioelectron, 2016, 83: 193–199. DOI:10.1016/j.bios.2016.04.075 |
| [60] | Wang DD, Jin Q, Zou LW, et al. A bioluminescent sensor for highly selective and sensitive detection of human carboxylesterase 1 in complex biological samples[J]. Chem Commun (Camb), 2016, 52: 3183–3186. DOI:10.1039/C5CC09874B |
| [61] | Ding LL, Tian ZH, Hou J, et al. Design and development of fluorescent probe substrates for carboxylesterase 1 using BODIPY as the basic fluorophore[J]. Acta Pharm Sin (药学学报), 2017, 52: 58–65. |
| [62] | Sato Y, Miyashita A, Iwatsubo T, et al. Simultaneous absolute protein quantification of carboxylesterases 1 and 2 in human liver tissue fractions using liquid chromatography-tandem mass spectrometry[J]. Drug Metab Dispos, 2012, 40: 1389–1396. DOI:10.1124/dmd.112.045054 |
| [63] | Hosokawa M, Endo T, Fujisawa M, et al. Interindividual variation in carboxylesterase levels in human liver microsomes[J]. Drug Metab Dispos, 1995, 23: 1022–1027. |
| [64] | Xu G, Zhang WH, Ma MK, et al. Human carboxylesterase 2 is commonly expressed in tumor tissue and is correlated with activation of irinotecan[J]. Clin Cancer Res, 2002, 8: 2605–2611. |
| [65] | Zhu HJ, Patrick KS, Yuan HJ, et al. Two CES1 gene mutations lead to dysfunctional carboxylesterase 1 activity in man:clinical significance and molecular basis[J]. Am J Hum Genet, 2008, 82: 1241–1248. DOI:10.1016/j.ajhg.2008.04.015 |
| [66] | Tarkiainen EK, Holmberg MT, Tornio A, et al. Carboxylesterase 1 c.428G > A single nucleotide variation increases the antiplatelet effects of clopidogrel by reducing its hydrolysis in humans[J]. Clin Pharmacol Ther, 2015, 97: 650–658. DOI:10.1002/cpt.v97.6 |
| [67] | Kubo T, Kim SR, Sai K, et al. Functional characterization of three naturally occurring single nucleotide polymorphisms in the CES2 gene encoding carboxylesterase 2(HCE-2)[J]. Drug Metab Dispos, 2005, 33: 1482–1487. DOI:10.1124/dmd.105.005587 |