 2018, Vol. 53
2018, Vol. 53



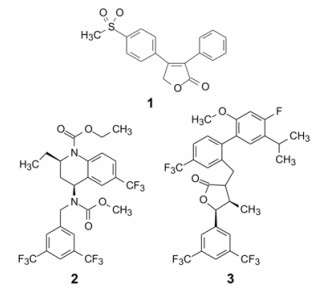
研发首创性药物需要发现靶标, 并将靶标确证贯穿于研发始终, 其中概念验证(proof of concept)于临床研究自不待言, 即使上市后在“真实世界”中应用, 仍在考量靶标的可药性(druggability), 成败教训不少。例如1999年上市的抗炎药罗非昔布(1, rofecoxib), 一度成为重磅药物, 后因心血管事件于2004年撤市, 并导致对靶标环氧合酶(COX)作彻底性质疑, COX-2虽躲过一劫, 却招致非甾体抗炎药(NSAID)和选择性COX2抑制剂的安全性作黑框警示。另一是胆固醇酯转运蛋白(CETP)抑制剂的研发, 辉瑞的托塞彻普(2, tolcetrapib)和默克的阿那彻普(3, anacetrapib)等候选物进入大规模的Ⅲ期临床, 都因心血管事件而终止, 损失惨重, 表明CETP缺乏可药性。然而羟基甲基戊二酸-辅酶A (HMG-CoA)还原酶却是研制降胆固醇药物的靶标, 在阻断胆固醇体内合成的级联反应的早期环节抑制六碳片段的合成转化, 竟无其他显著的不良反应, HMG-CoA还原酶证明是优质靶标。
|
研制跟随性药物是首创性药物的后续与补充, 后来者往往居上, 成为新药创制的重要内容。由于作用靶标证实具备可药性, 跟随性研发无需作靶标确证, 风险也相对较低, 难度似乎较小。这在20世纪80年代有明显优势, 因为那时的新药创制理念尚不充实, 首创药物在药理活性和(或)成药性(drug likeness)的优化有失充分, 客观上给跟随性研究留有超越的空间, 导致同类首创药物(first in class)较少是同类最优药物(best in class)。例如降胆固醇的第五个上市药物阿托伐他汀优于首创药洛伐他汀、抗菌药左氟沙星优于首创的诺氟沙星等。
然而本世纪以来首创药物在活性(强度和选择性)和成药性(药代、安全性和物化性质等)的优化相对比较充分, 专利覆盖范围也广泛, 跟随性研发空间变小, 加之药监部门要求后继者优于(至少不劣于)首创药, 超越难度加大。另一个特点是由于研发周期长, 首创药专利到期与后继药物上市时间相近, 销售与市场风险加大。即使都是以首创的方式完成并相继问世的药物, 竞争也会很激烈。例如一批降血糖药物2型钠葡萄糖共转运蛋白(SGLT2)抑制剂短时间内密集上市, 坎格列净(4, canagliflozin, 2013年)、达格列净(5, dapagliflozin, 2014年)和艾帕列净(6, empagliflozin, 2014年)都是以首创进行研发。它们的药效与化学类型相似, 都是以天然活性物质根皮苷为先导物研发的。
|
真正意义上的跟随性创新是在首创研发进入临床研究前后启动的项目, 风险相对降低, 但由于滞后时间较长, 后置的风险需要性质优胜以规避、超越和弥补先驱药物的不足。为此, 在研发策略上应作考究。笔者认为可从两方面切入, 一是从小分子化合物入手, 有针对性的作结构修饰(例如骨架迁越和修饰), 在安全、疗效或方便使用等某一(些)目标进行优化, 以优于已有的同类药物; 另一是从大分子靶标入手, 根据深化的作用机制或靶标的变异创制新的化学结构, 基于微观的结构特征或反应机制研发小分子, 作互补性构建。两种策略是密切相连的, 为方便起见而分开叙述。本文拟就上市的一些跟随性药物, 阐述研发的一些要旨和特征, 在众多成功的跟随性药物中, 只能挂一漏万。
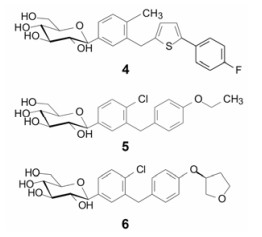
2 化学结构的深化修饰 2.1 GLP-受体激动剂的长效化2型糖尿病患者需要长期服药, 若药物作用时间长, 可降低用药频次, 为此可用长效剂型, 也可变换药物的化学结构而长效化。治疗2型糖尿病的胰高血糖素样肽(GLP-1)受体激动剂可改善β细胞功能, 减肥和降低收缩压, 是控制血糖减轻体重的药物, GLP-1的活性区段是三十一肽(7), 二肽基肽酶4 (DPP-4)的水解位点是Ala8-Glu9肽键, 肽类GLP-1受体激动剂要克服DPP-4的水解作用。
首创药物艾塞那肽(8, exenatide)是天然多肽毒蜥外泌肽(exendin-4)未经改构的合成品, 皮下注射半衰期t1/2 = 2~3 h, 礼来公司于2005年上市, 皮下注射给药, 每日两次, 这对患者是不便的。其跟随性药物采用的是结构修饰使长效化的策略。
2010年由诺和诺德公司研发上市的利拉鲁肽(9, liraglutide)是对GLP-1 (7-37)的修饰, 将Lys34变换为Arg34, 并在Lys26的ξ氨基经另一赖氨酸连接出C16脂肪酸链, 该脂质性侧链有利于同白蛋白非极性区结合, 白蛋白具有结合并运载内源性物质的功能。在血浆中由于利拉鲁肽与白蛋白结合, 提高了对DPP-4和内切酶的稳定性, 半衰期增长, t1/2 = 11~15 h, 延长了在血浆中的存留时间并降低被肾脏的清除, 利拉鲁肽皮下注射, 每日一次[1]。
|
礼来公司研发的度拉鲁肽(dulaglutide)于2004年上市, 是GLP-1 (7-37)的类似物与单抗IgG4的Fc片段的融合蛋白, 长效性进一步提高, 每周一次皮下注射可控制血糖水平。不足之处是其降低体重作用弱于利拉鲁肽[2]。
诺和诺德公司上市利拉鲁肽后, 进而研制一周给药一次的长效激动剂, 将GLP-1 (7-37)修饰为Aib8 (α-氨基丁酸)和Arg34-GLP-1 (8-37)多肽, 以提高与受体的结合力和稳定性, 再用不同长度的乙二醇-氨基乙醇醚为连接基, 将C18~C22二羧酸与Lys26残基相连, 最终优化出含有与白蛋白强效结合的疏水性侧链的塞马鲁肽(10, samaglutide), 表 1列出了塞马鲁肽与利拉鲁肽的受体激动活性和药代性质, 虽然塞马鲁肽体外活性低于利拉鲁肽3倍, 但对Goettingen微型猪的药代明显占优。例如皮下注射后生物利用度提高30%, 体内平均存留时间(MRT)塞马鲁肽为利拉鲁肽的3倍。塞马鲁肽于2017年10月FDA批准上市, 每周皮下注射一次, 具有控制血糖和减肥的药效, 同时可降低心血管事件风险(MACE)[3]。
| 表 1 塞马鲁肽与利拉鲁肽的体外活性与药代(微型猪)主要参数比较 |
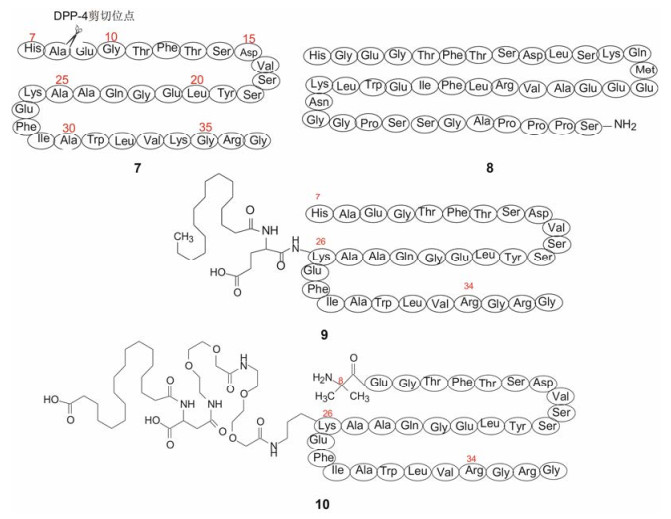
毒蕈碱乙酰胆碱受体3 (简称M3受体)高表达于支气管平滑肌, 激动M3受体可引起支气管收缩, 抑制M3受体是治疗慢性阻塞性肺病(COPD)和哮喘的环节。M3的同源蛋白M2受体高表达于心脏, 对M2和M3选择性不强的抑制剂会发生心血管的不良反应。此外, COPD作为慢性病需长期应用, 长效有利于患者使用。
季铵类首创M3拮抗剂异丙托溴铵(11, ipratro-pium bromide)于1986年FDA批准在美国上市, 吸入治疗COPD, 每日吸入4~6次, 每次40~80 mg。后继的选择性拮抗剂噻托溴铵(12, tiotropium bromide)和阿地溴铵(13, aclidinium bromide)是FDA在2012年批准上市的。这两个跟随性药物结构的特点是增强了分子的疏水性, 并提高了对M3受体的选择性抑制作用, 是由于对M3和M2的结合动力学性质有显著区别的缘故。
|
体外评价配体与受体的可逆性结合强度(Kd、IC50)是以靶标为核心初步评价受试物活性的指标, 但Kd、IC50不能完全反映在体内与受体的结合状态, 因为复合物的生成及离解速率有快慢之分, 配体与受体在体内的开放系统中, 药物浓度不断变化。若配体在受体结合部位驻留时间长, 即使游离配体水平已低于Kd值, 仍占据受体而保持药理活性, 其根源在于复合物的离解速率低。受体蛋白通过构象选择或经诱导契合与配体结合, 蛋白发生构象变化, 构象变化越大, 复合物离解回复到游离状态越难, 需要越过较高的能垒, 导致离解减慢[4]。噻托溴铵是慢离解的M3受体阻断剂。吸入治疗, 每日一次, 每次18 mg, 显著优于异丙托溴铵。
表 2列出了异丙托溴铵和噻托溴铵的生物学参数。两个药物对M3和M2受体结合常数Kd虽然相差10倍, 但比值相同, 倘只从热力学性质分析, 二者对M3的选择性相同。但噻托溴铵对M3的离解速率(koff)显著低于M2, 也低于异丙托溴铵的离解速率, 因而在M3受体上的驻留时间(residence time)显著延长, 停留并结合在M3的时间长, 从而用药剂量和频度少于异丙托溴铵, 也因此减低了对M2受体的脱靶作用[5]。阿地溴铵的长效性也是由于慢离解速率的缘故。如今新药研究中已注重评价化合物结合动力学性质(特别是离解速率常数)[6], 预测在体内的长效性。
| 表 2 异丙托溴铵和噻托溴铵的结合动力学性质、选择性和用法 |
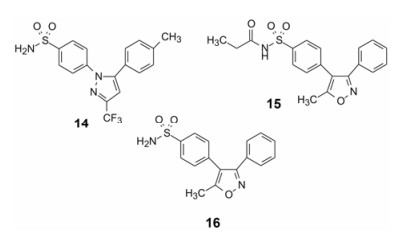
选择性环氧合酶2 (COX-2)抑制剂塞来昔布(14, celecoxib)是辉瑞公司研发的首创性抗炎药, 上市于1999年, 获得巨大成功。辉瑞的后继产品帕瑞昔布(15, parecixb)于2002年上市, 以钠盐形式注射用药。帕瑞昔布在体内经代谢水解生成伐地昔布(16, valdecoxib)起效[7], 所以是前药。分子中的磺酰氨基经丙酰化, 氨基呈酸性(双酰基拉电子), 可制成可溶性钠盐。释放出的伐地昔布与塞来昔布有相同的药效团特征, 只是咪唑环骨架变为异噁唑环。作为唯一注射用的COX-2抑制剂, 帕瑞昔布改变了适应症, 用于解除围术期中度或重度疼痛, 例如用于牙科、骨科和妇科手术的止痛, 避免或减少了吗啡的使用[8]。
|
第一个治疗艾滋病的HIV蛋白酶抑制剂茚地那韦自1995年上市以来, 到2006年地瑞那韦获得批准, 之间有多个“那韦”药物上市, 这些抑制剂的结构类型都是模拟蛋白底物被酶水解时的过渡态, 因而是一类过渡态类似物。虽然与酶的结合模式相同, 但过渡态结构所处的分子环境不同, 因而与酶结合强度和热力学性质有明显差异。其中安普那韦(17, amprenavir)是由GSK公司研发, 于1999年上市的, 7年后Tibotec公司研发的地瑞那韦(18, darunavir)与其结构相似, 是在安普那韦四氢呋喃环上再并合一个环成为双环, 抑制蛋白酶活性因此提高了近百倍。结构生物学研究揭示, 新并合环的氧原子与Asp30'和Asp29'发生两个氢键结合, 形成的氢键网络增高了与靶标的结合力, 如图 1所示。此外, 热力学分析提示, 氢键提供的结合能(ΔG)主要是焓贡献(ΔH), 由于焓比熵(-TΔS)更显示结合的特异性, 地瑞那韦结合强度和特异性更强。表 3列出的数据表明, 安普那韦(17)的焓熵贡献各占一半, 而地瑞那韦(18)则主要是焓贡献[9]。

|
图 1 地瑞那韦与HIV蛋白酶复合物的结合模式。虚线代表氢键结合, 圆点为参与复合物生成的结构水 |
| 表 3 安普那韦和地瑞那韦的活性和热力学参数 |
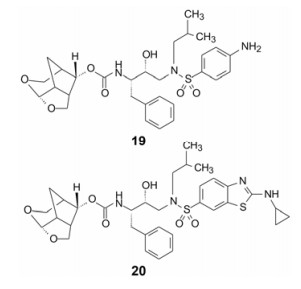
地瑞那韦的发明者基于晶体结构的结合模式, 认为在双环端的疏水结合腔仍有空间, 从而设计了立体冠状的含氧三环结构化合物19, 以代替地瑞那韦的双环片段, 19的活性有所提高(抑制病毒感染细胞的IC50 = 3.3 nmnol·L-1, 地瑞那韦IC50 = 3.8 nmnol·L-1), 是由于冠状的含氧片段不仅保持了氢键网络, 还有利于同leu47、Val32和Leu76构成腔穴发生范德华和疏水性结合, 如图 2a所示, 而地瑞那韦(图 2b)该处未发生疏水性结合。进而将另端的苯胺片段用氨基苯并噻唑替换, 化合物20的活性提高了近10倍(IC50 = 0.39 nmnol·L-1), 20还对耐受地瑞那韦的病毒感染有强抑制活性[10]。

|
图 2 a.化合物19的冠醚与酶的结合方式; b.地瑞那韦的双四氢呋喃与蛋白酶的S2结合方式 |
|
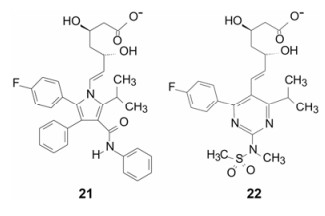
增加氢键结合和范德华作用比疏水性结合具有更强的特异性, 据此, 跟随性药物可提高药理作用的特异性, 也避免代谢的复杂性。阿托伐他汀(21, atorvaststin)和瑞舒伐他汀(22, rosuvaststin)与HMG-CoA还原酶的结合是这方面的实例。21和22是第5和第6个上市的“他汀”类药物, 临床应用和市场份额比4个前驱药物优胜, 重要原因是结构因素具有特点。在21和22的分子的下半部的疏水片段上分别引入了酰胺基和磺酰基片段, 增加了氢键的结合, 该特异性结合反映在21和22与酶结合能的组成上, 焓贡献是主要驱动力, 而前4个他汀则是以熵驱动为主的结合[11]。
|
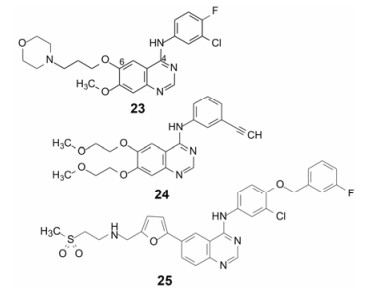
2003年FDA批准阿斯利康公司的吉非替尼(23, gefetinib)和罗氏公司2004年上市的厄罗替尼(24, erlotinib)都是酪氨酸蛋白激酶EGFR抑制剂, 结合于EGFR酪氨酸激酶的ATP结合位点, 治疗非小细胞肺癌, 批准时间相近, 在独立进行的意义上都可视作首创性研发, 二者分子骨架相同, 虽然助溶基团不同, 也都连接于喹唑啉的6位。
GSK于2007年上市的拉帕替尼(25, lapatinib)也作用于酪氨酸激酶, 是EGFR和HER2/neu双靶标抑制剂, 治疗HER2阳性的乳腺癌。25也具有相同的4-苯胺基喹唑啉骨架和6位助溶基团, 但作为跟随性药物, 区别在于将23和24的苯胺环上简单取代基变为经氧亚甲基再连接一取代的苯环。分子模拟显示, 新加的取代苯基诱导使得酶的C-螺旋位置改变, 苯环伸入到深部被打开的腔内, 这个腔在apo-EGFR和24的复合物晶体结构中不存在。这样, 诱导契合适配于酶结合部位的空间, 增添了新的结合因素。新增的结合腔在结合动力学性质上有所不同。24与EGFR的复合物晶体结构与apo-EGFR基本相同, 并且有较快的离解速率, 复合物的半衰期t1/2 = 10 min (驻留时间RT = 14 min), 比较快速的离解说明厄罗替尼结合或离开EGFR活性部位时, 蛋白的构象无需发生重大的变化。而25的复合物的t1/2 = 300 min (RT = 433 min), 为了离解成游离状态, 需要改变EGFR的构象, 因而成为慢离解性药物[12]。
|
后继跟随的一系列EGFR激酶抑制剂中的来那替尼(26, neratinib)为HER2激酶抑制剂, 2017年上市, 临床治疗HER2呈阳性的乳腺癌。来那替尼的化学结构具有优化组合的特征, 将已证明有效的片段或因素整合到一个分子结构中。分子模拟结果是: ①母核用3-氰基喹啉替换喹唑啉环。3-氰基代替了喹唑啉的N3, N3与水分子经氢键介导与Ser783的OH形成氢键, 而3-氰基则直接与OH生成氢键(2.76 Å), 减少了熵损伤(变三元为二元组合); ② 6位氨基经4-二甲胺基丁-2-烯酸酰化, 成为弱麦克尔基团, β碳与酶开口处的Cys805的巯基相距3.43 Å,可发生亲电加成反应, 巯基被烷基化, 而且末端的二甲胺基经分子内催化促进了该共价键的生成; ③吡啶甲氧基处于上述拉帕替尼相似的疏水腔内[13]。
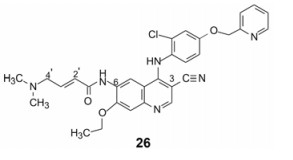
|
氘代丁苯那嗪(27, deutetrabenazine)是2017年FDA批准Auspex公司上市的, 治疗中枢神经性疾病亨廷顿氏病等舞蹈病, 也是FDA批准的全球首个氘代药物。丁苯那嗪是20世纪50年代已知的化合物, 直到2008年FDA批准治疗亨廷顿氏病。其作用机制是靶向抑制囊泡单胺转运蛋白2 (vesicular monoamine transporter 2, VMAT-2), 阻断并抑制单胺类神经递质的储存, 减少单胺对突触小泡的摄取。口服丁苯那嗪在肝脏被CYP2D6氧化脱甲基, 首过效应导致生物利用度低。代谢环节是甲氧基的C-H被氧化。由于C-D键强于C-H, 利用动力学同位素效应(kH/kD = 6.5), 研制了氘代丁苯那嗪, 只在代谢位点作氘代, 并用光学活性的R, R-异构体, 提高了代谢稳定性, 降低了用药剂量和频次[14]。
我国的泽璟生物公司研发的多纳非尼(28, donafenib)目前处于Ⅲ期临床阶段, 是拜耳首创的索拉非尼的氘代物。
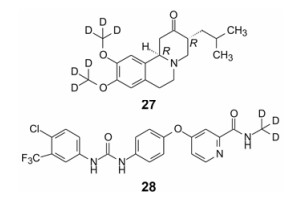
|
2004年礼来公司上市的抗肿瘤药物培美曲塞(29, pemetrexed)是与普林斯顿大学Taylor等共同研制的, 为甲氨蝶呤(30, methotrexate)的跟随性药物, 同为叶酸抗代谢物。1950年上市的甲氨蝶呤开始了现代意义的肿瘤化学治疗, 至今仍在临床应用。甲氨蝶呤作为辅酶叶酸(31, folic acid)的抗代谢物, 是二氢叶酸还原酶(DHFR)抑制剂, DHFR催化二氢叶酸还原成四氢叶酸(THF), 后者作为辅酶在细胞中的一碳转化反应起重要作用, 参与体内胸苷酸、嘌呤、蛋氨酸等的de novo合成以及丝氨酸-甘氨酸的相互转化。
化学家Taylor从分析辅酶的功能入手, 发现叶酸参与胸苷酸合成酶(TS)和二氢叶酸还原酶(DHFR)的生化反应, TS和DHFR都是合成DNA前体物胸腺嘧啶脱氧核苷(dTMP)的关键酶, 在细胞增殖中起重要作用。同时抑制这两个酶系, 可提高对癌的化疗效果。另一酶系叶酸聚谷氨酸合成酶(FPGS)是将进入细胞内的叶酸发生聚谷氨酸化而起效, 抗代谢物若成为PFGS底物(Km值小), 容易在此环节上发挥抗癌作用。因而研制新叶酸抗代谢物的目标是同时作用于DHFR、TS和FPGS这三个酶系, 在多靶标的作用中提高对癌细胞的选择性, 避免甲氨蝶呤的不良反应。
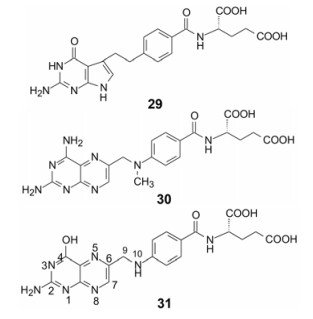
|
Taylor分析了二氢叶酸和四氢叶酸都具有蝶啶-对氨基苯甲酸-谷氨酸的结构, 区别只是N5和N10的连接基团, 因而设想变换的位点应集中在N5和N10处。为此, 围绕着N5和(或) N10的原子和环-链的变换, 探索化合物对上述3种酶的抑制作用。经构效关系(SAR)和试错反馈(trial and error), 凭借娴熟的杂环化学, 合成了700多个化合物, 成功地研制出培美曲塞[15-20]。
表 4对比了培美曲塞与甲氨蝶呤体外活性的数据。培美曲塞对FPGS一级速率常数相对值为400(以叶酸为100), 甲氨蝶呤为0.9, 显著强于甲氨蝶呤, 提示培美曲塞在细胞内容易发生聚谷氨酸化, 这一环节是抗代谢物呈现活性的关键步骤。后经化学合成的五聚谷氨酸培美曲塞对离体TS、氨基咪唑酰胺核苷酸甲基转移酶(AICARFT)和四氢叶酸合成酶都呈现强抑制活性[21]。
| 表 4 培美曲塞与甲氨蝶呤的体外活性比较 |
培美曲塞与顺铂合用静脉滴注治疗恶性胸膜间皮瘤。作为甲氨蝶呤的第一个跟随性药物与之相隔半个世纪, 是从大学的学术研究(杂环化学的合成和叶酸抗代谢物的功能)转化到药厂开发研究的成功范例。
3.2 针对发生变异的肿瘤靶标诺华公司2001年上市的伊马替尼(32, imatinib)治疗慢性粒细胞白血病(CML), CML由于是血液干细胞过分增殖, 第22号和第9号染色体交互易位, 导致22号染色体变短(称作费城染色体), 成为癌症与基因变异之间关系的最早的证据。染色体易位的分子基础是Bcr-Abl编码的蛋白酪氨酸激酶过度活化所致。伊马替尼抑制Abl酪氨酸激酶活性, 阻止ATP对蛋白的磷酸化, 由于针对的是CML特异基因的表达产物, 成为精准设计的高度选择性药物, 也因此开创了分子靶向药物的新领域[22]。
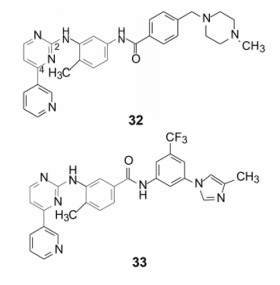
|
伊马替尼获得了巨大成功, 但在持续用药中出现耐药性。重要原因是酶的活性中心氨基酸残基发生变异, 例如门户残基Thr315变异成Ile315 (T315I), T315I突变的后果是氨基酸侧链由CH(CH3)OH变成CH(CH3)CH2CH3, 不仅体积变大, 空间上阻碍了伊马替尼的进入, 而且失去了形成氢键结合的能力(羟基O为氢键接受体)。然而这种变异并不妨碍Abl与ATP的结合和磷酸化的发生。此外, 还发生Glu255突变为Val255, 也失去了形成氢键能力, 使活化环套的DFG-in处于更加稳定的状态, 反而使伊马替尼结合所需的DFG-out构象减少或不稳定了[23]。
诺华公司为了克服伊马替尼的耐药性, 于2007年研制出后继的药物尼洛替尼(33, nilotinib), 是基于伊马替尼与Abl激酶复合物晶体结构, 利用药物化学和构效关系设计出来的。直观看二者化学结构相似, 都有4-吡啶基-2-苯胺基嘧啶结的骨架, 其实经Daylight-fingerprint-Tanimoto模型计算, 32和33的相似性系数只有0.6 (全同为1.0, 完全不同为0)。主要区别是酰胺键的方向不同(由胺酰键变为酰胺键)和末端苯环上取代基的差异, 使得尼洛替尼分子构象、拓扑形状和基团结合的模式发生了变化。例如, 尼洛替尼的酰胺基使得Abl激酶的Glu286为了与NH结合而移位, 从而更加稳定了DFG-out构象, 并腾出了空间有利于咪唑基的进入。这样, 不仅克服了耐药性, 对酶和转染细胞的抑制活性提高了10~30倍[24, 25]。
尼罗替尼的结构设计没有利用酶发生的T315I变异的因素。ARIAD公司研制对突变的Bcr-Abl激酶抑制剂, 结构类型是嘌呤N9经乙烯基与苯甲酰胺连接的骨架, 如化合物34, 分子模拟显示乙烯基处于变异的Ile315处, 如图 3所示。未发生变异这个位置为羟甲基, 伊马替尼的嘧啶2-氨基与之形成氢键。当变异成异丁基, 体积变大, 而且失去氢键结合能力, 因此伊马替尼的-NH-对T315I失去作用, 成为多余。然而乙烯基与异丁基发生空间障碍, 不利于结合, 遂变成体积小的乙炔基, 解除了位阻, 有利于结合。进一步变换其他结构片段, 最后优化出选择性抑制T315I突变型Bcr-Abl抑制剂帕那替尼(35, ponatinib), 于2012年上市, 治疗伊马替尼耐药的慢性粒细胞白血病。图 4是帕那替尼的对接图[26]。

|
图 3 化合物34与T315I酶对接图 |

|
图 4 帕那替尼与T315I酶对接图 |
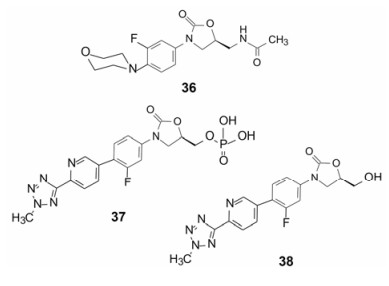
首创性药物利奈唑胺(36, linezolid)是继20世纪30年代的磺胺、40年代青霉素为代表的抗生素和20世纪80年代的喹诺酮之后的新型结构(噁唑烷酮)抗菌药。利奈唑胺是细菌蛋白合成的抑制剂, 作用环节是在核糖体上阻断mRNA翻译成蛋白的初始阶段, 结合部位是核糖体50S亚基的A位点。由于抑菌活性强而广谱, 吸收好(口服生物利用度F = 100%), 鲜有跟随的噁唑烷酮上市, 直到2014年第二个跟随性药物特地唑胺(37, tedizolid)成功。特地唑胺是以磷酸酯为前药, 可口服或注射用药, 在血浆中被磷酸酶水解掉磷酸基成活性形式(38, TR-700)。对耐受甲氧西林的革兰阳性菌的抑制作用强于利奈唑胺4~16倍。日用一次[27]。
|
特地唑胺的研制路径是在分析众多构效关系的基础上成功的, 图 5是噁唑烷酮类的构效关系, 通过对C和D环的加入和变换, 发现C和D环的存在具有广谱抗菌特征, 优化出2-吡啶基连接的推电子片段甲基四氮唑。分子模拟显示, 38的C和D环结合于转肽酶的活性部位的保守肽段, 更重要的是, 噁唑烷酮的5S-位羟甲基替换了利奈唑胺的乙酰胺甲基, 提高了对耐药菌的敏感性。这是因为利奈唑胺耐药菌的23S rRNA碱基A2503在C8位发生甲基化, 后者因与利奈唑胺的乙酰氨基有位阻效应而降低结合, 38的羟甲基则解除了该空间障碍, 因而对耐药株具有敏感性[28]。

|
图 5 噁唑烷酮类抗菌作用的构效关系 |
研发跟随性药物是首创药物的必然后续和补充, 但得有度, 体现在有特点以优于(或不劣于)首创药物, 而且数量上也有限度, 因市场容量有限。精准医疗要求精确创制特异的药物, 因而跟随性药物要设定明确的目标, 有针对性。在药效、药代、安全性、物化性质(往往影响给药途径、剂量和频次)等无论哪一方面优胜于已有药物, 就有可取之处。克服化疗(感染和肿瘤)药物的耐药性的新药研发, 是永恒的主题, 跟随性的创新意义重大。以靶标为核心的新药创制, 由分子→细胞→动物模型→患者的转化中有许多不确定因素, 始料未及的成功有幸运因素。激酶抑制剂的抗肿瘤药物的适应症是“打哪儿指哪儿”, 而难以“指哪儿打哪儿”, 有宿命意味。例如第二个选择性雌受体调节剂(SERM)雷洛昔芬的适应症是骨质疏松症, 不同于首创药他莫昔芬治疗乳腺癌, 虽然二者的药效团特征相同。在药代的ADME中最难确定的是分布D, 药物的体内分布在很大程度上决定了选择性、安全性和适应症, 所以在这个意义上, 研发跟随性药物仍有很大的空间。
| [1] | Araki E, Inagaki N, Tanizawa Y, et al. Efficacy and safety of once-weekly dulaglutide in combination with sulphonylurea and/or biguanide compared with once-daily insulin glargine in Japanese patients with type 2 diabetes:a randomized, open-label, phase Ⅲ, non-inferiority study[J]. Diabetes Obes Metab, 2015, 17: 994–1002. DOI:10.1111/dom.12540 |
| [2] | Dungan KM, Povedano ST, Forst T, et al. Once-weekly dulaglutide versus once-daily liraglutide in metformin-treated patients with type 2 diabetes (AWARD-6):a randomised, open-label, phase 3, noninferiority trial[J]. Lancet, 2014, 384: 1349–1357. DOI:10.1016/S0140-6736(14)60976-4 |
| [3] | Lau J, Bloch P, Schaeffer L, et al. Discovery of the once-weekly glucagon-like peptide-1(GLP-1) analogue semaglu-tide[J]. J Med Chem, 2015, 58: 7370–7380. DOI:10.1021/acs.jmedchem.5b00726 |
| [4] | Copeland RA. Conformational adaptation in drug-target interactions and residence time[J]. Future Med Chem, 2011, 3: 1491–1501. DOI:10.4155/fmc.11.112 |
| [5] | Barnes PJ. The pharmacological properties of tiotropium[J]. Chest, 2000, 117(suppl): 63–66. |
| [6] | Cusack KP, Wang Y, Hoemann MZ, et al. Design strategies to address kinetics of drug binding and residence time[J]. Bioorg Med Chem Lett, 2015, 25: 2019–2027. DOI:10.1016/j.bmcl.2015.02.027 |
| [7] | Talley JJ, Bertenshaw SR, Brown DL, et al. N-[(5-Methyl-3-phenylisoxazol-4-yl)-phenyl] sulfonyl] propanamide, sodium salt, parecoxib sodium:a potent and selective inhibitor of COX-2 for parenteral administration[J]. J Med Chem, 2000, 43: 1661–1663. DOI:10.1021/jm000069h |
| [8] | Cheer SM, Goa KL. Parecoxib (parecoxib sodium)[J]. Drugs, 2001, 61: 1133–1141. DOI:10.2165/00003495-200161080-00010 |
| [9] | Tie YPI, Boross YF, Wang L, et al. High resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multidrug-resistant clinical strains[J]. J Mol Biol, 2004, 338: 341–352. DOI:10.1016/j.jmb.2004.02.052 |
| [10] | Ghosh AK, Rao KV, Nyalapatla PR, et al. Design and development of highly potent HIV-1 protease inhibitors with a crown-like oxotricyclic core as the P2-ligand to combat multidrug-resistant HIV variants[J]. J Med Chem, 2017, 60: 4267–4278. DOI:10.1021/acs.jmedchem.7b00172 |
| [11] | Sarver RW, Bills E, Bolton G, et al. Thermodynamic and structure guided design of statin based inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase[J]. J Med Chem, 2008, 51: 3804–3813. DOI:10.1021/jm7015057 |
| [12] | Wood ER, Truesdale AT, McDonald OB, et al. A unique structure for epidermal growth factor receptor bound to GW572016(lapatinib). Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells[J]. Cancer Res, 2004: 6652–6659. |
| [13] | Tsou HR, Overbeek-Klumpers EG, Hallett WA, et al. Optimization of 6, 7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity[J]. J Med Chem, 2005, 48: 1107–1131. DOI:10.1021/jm040159c |
| [14] | Citrome L. Breakthrough drugs for the interface between psychiatry and neurology[J]. Int J Clin Pract, 2016, 70: 298–299. DOI:10.1111/ijcp.2016.70.issue-4 |
| [15] | Taylor EC, Harrington PJ, Fletcher SR, et al. Synthesis of the antileukemic agents 5, l0-dideazaaminopterin and 5, l0-dideaza-5, 6, 7, 8-tetrahydroaminopterin[J]. J Med Chem, 1985, 28: 914–921. DOI:10.1021/jm00145a012 |
| [16] | Taylor EC, Hamby JM, Shih C, et al. Synthesis and antitu-mor activity of 5-deaza-5, 6, 7, 8-tetrahydrofolic acid and its N10-substituted analogues[J]. J Med Chem, 1989, 32: 1517–1522. DOI:10.1021/jm00127a019 |
| [17] | Taylor EC, Harrington PM, Shih C. A facile route to "open chain" analogues of DDATHF[J]. Heterocycles, 1989, 28: 1169–1178. DOI:10.3987/COM-88-S146 |
| [18] | Taylor EC, Gillespie P, Patel M. Novel 5-desmethylene analogues of 5, 10-dideaza-5, 6, 7, 8-tetrahydrofolic acid as potential anticancer agents[J]. J Org Chem, 1992, 57: 3218–3225. DOI:10.1021/jo00037a047 |
| [19] | Shih C, Gossett LS, Worzalla JF, et al. Synthesis and biological activity of acyclic analogues of 5, 10-dideaza-5, 6, 7, 8-tetrahydrofolic acid[J]. J Med Chem, 1992, 35: 1109–1116. DOI:10.1021/jm00084a016 |
| [20] | Taylor EC. The discovery of Alimta (pemetrexed)[M]//. Fischer J, Rotella DP. Successful Drug Discovery. Vol 1. Weinheim, Germany: Wiley-VCH, 2015: 157-180. |
| [21] | Taylor EC, Kuhnt D, Shih C, et al. A dideazatet-rahydrofolate analogue lacking a chiral center at C-6, N-[4-[2-(2-amino-3, 4-dihydro-4-oxo-7H-pyrrolo[2, 3-d]pyrimidin-5-yl)ethyl]benzoyl]-L-glutamic acid, is an inhibitor of thymidylate synthase[J]. J Med Chem, 1992, 35: 4450–4454. DOI:10.1021/jm00101a023 |
| [22] | Zimmermann J, Buchdunger E, Mett H, et al. Potent and selective inhibitors of the Abl-kinase phenyl-amino-pyrimidine (PAP) derivatives[J]. Bioorg Med Chem Lett, 1997, 7: 187–192. DOI:10.1016/S0960-894X(96)00601-4 |
| [23] | Cowan-Jacob SW, Guez V, Fendrich G, et al. Imatinib (STI571) resistance in chronic myelogenous leukemia:molecular basis of the underlying mechanisms and potential strategies for treatment[J]. Mini-Rev Med Chem, 2004, 4: 285–299. DOI:10.2174/1389557043487321 |
| [24] | Weisberg E, Manley PW, Breitenstein W, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl[J]. Cancer Cell, 2005, 7: 129–141. DOI:10.1016/j.ccr.2005.01.007 |
| [25] | Manley P, Stiefl N, Cowan-Jacob S, et al. Structural resem-blances and comparisons of the relative pharmacological properties of imatinib and nilotinib[J]. Bioorg Med Chem, 2010, 18: 6977–6986. DOI:10.1016/j.bmc.2010.08.026 |
| [26] | Huang WS, Metcalf CA, Sundaramoorthi R, et al. Discovery of 3-[2-(imidazo[1, 2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl)-methyl]-3-(trifluoromethyl)phenyl} benzamide (AP24534), a potent, orally active pan-inhibitor of breakpoint cluster region-abelson (BCR-ABL) kinase including the T315I gatekeeper mutant[J]. J Med Chem, 2010, 53: 4701–4719. DOI:10.1021/jm100395q |
| [27] | Schaadt R, Sweeney D, Shinabarger D, et al. In vitro activity of TR-700, the active ingredient of the antibacterial prodrug TR-701, a novel oxazolidinone[J]. Antimicrob Agents Chemother, 2009, 53: 3236–3239. DOI:10.1128/AAC.00228-09 |
| [28] | Locke JB1, Finn J, Hilgers M, et al. Structure-activity rela-tionships of diverse oxazolidinones for linezolid-resistant Staphylococcus aureus strains possessing the cfr methyltransferase gene or ribosomal mutations[J]. Antimicrob Agents Chemother, 2010, 54: 5337–5343. DOI:10.1128/AAC.00663-10 |