 2017, Vol. 52
2017, Vol. 52



2. 山西大学 化学化工学院, 山西 太原 030006;
3. 山西大学 地产中药功效物质研究与利用山西省重点实验室, 山西 太原 030006;
4. 中国医学科学院药物研究所, 北京 100050
2. College of Chemistry and Chemical Engineering, Shanxi University, Taiyuan 030006, China;
3. Shanxi Key Laboratory of Active Constituents Research and Utilization of TCM, Shanxi University, Taiyuan 030006, China;
4. Institute of Materia Medica, Chinese Academy of Medical Sciences, Beijing 100050, China
抑郁症是一种慢性复发性情绪障碍, 是全球常见的疾病之一。多因生活节奏快引起的慢性应激使神经和免疫功能发生变化, 进而导致抑郁症[1]。在临床中, 抑郁症患者常表现出注意力不集中、思考能力下降、失眠、食欲缺乏等症状[2]。目前, 抑郁症的发病机制尚未明确, 但现有的抗抑郁药物可明显改善抑郁症状。通过明确抗抑郁药物的体内代谢过程, 明确代谢途径、代谢酶及其代谢物的药理毒理性质, 进而预测药物的体内代谢特征。进一步研究药物与酶的相互作用, 推测药物对酶的活性是否有影响, 避免不良反应的发生。同时, 在体内生物转化过程中产生活性较高的代谢物, 也是新型候选化合物的来源与途径[3]。本文选取了临床中常用的抗抑郁药物, 综述其体内代谢过程, 为抗抑郁新药的研发提供思路。
1 抑郁症的发病机制及抗抑郁药物的发展抑郁症是一种涉及多种神经递质、脑区及环路的疾病, 且脑内其他生化物质也参与了抑郁症的病理学过程。虽然, 抑郁症在分子和细胞方面的发病机制仍有待评估, 但单胺类神经递质、谷氨酸信号及各种生长因子已被证实与抑郁症有关[4]。
目前市售有明确疗效的抗抑郁药物多作用于神经末梢突触部位, 通过调节单胺类神经递质在突触的再摄取过程发挥治疗作用。
根据抗抑郁药物的作用机制及被开发时间, 临床上将常用的抗抑郁药物分为如下几类, 即第一代抗抑郁药物:单胺氧化酶抑制剂(monoamine-oxidase inhibitors, MAOIs)和三环类抗抑郁药物(tricyclic antidepressants, TCAs); 第二代抗抑郁药物:选择性5-羟色胺再摄取抑制剂(selective serotonin reuptake inhibitors, SSRIs); 第三代抗抑郁药物: 5-羟色胺和去甲肾上腺素双重再摄取抑制剂(serotonin and noradrenaline reuptake inhibitors, SNRIs)、去甲肾上腺素再摄取抑制剂(noradrenaline reuptake inhibitors, NDRIs)及去甲肾上腺素和5-羟色胺能抗抑郁剂(noradrenergic and specific serotonergic antidepressants, NaSSA)等[5]。另外, 由于临床使用化学药物时常伴有不同程度不良反应发生, 因此具有抗抑郁作用的单一成分天然药物或复方中药在治疗抑郁症中也逐渐被广泛使用[6]。目前, 已有明确疗效的抗抑郁天然药物及中药复方主要有圣约翰草[7]、莉芙敏(莉芙敏片)[8]及舒肝解郁胶囊等[9]。
2 抗抑郁药物的代谢 2.1 MAOIs的代谢第一代抗抑郁药物MAOIs代表性药物吗氯贝胺, 通过抑制线粒体中单胺氧化酶(MAO), 减低体内单胺类物质的氧化脱氨而发挥药效, 但在随后的研究及临床用药过程中发现对MAOs的抑制具有不可逆转性[10]。另一种MAOIs代表药物苯乙肼, 在肝脏中能被MAO氧化, 也对MAO具有不可逆转的抑制作用[11]。因此, 目前临床用药中已不再使用这类药物。
2.2 TCAs的代谢TCAs也为第一代抗抑郁药物, 通过抑制去甲肾上腺素和5-羟色胺的再摄取而发挥疗效。典型的TCAs类药物阿米替林(amitriptyline), 生物利用度良好, 口服后能被吸收并迅速分布于不同的组织器官中。在代谢过程中, 阿米替林在CYP2C19作用下发生N-去甲基化反应, 生成活性代谢产物去甲替林(nortriptyline, 被开发成商品名为Nortrilen®的抗抑郁药物[12]), 随后经CYP2D6代谢, 生成10位羟基化产物[13], 其原形药物及代谢产物主要通过肾排泄途径排出体外[14], 其代谢过程见图 1。但在临床治疗过程中发现, 此类药物对受体的阻滞导致心脏及中枢神经系统的不良反应强, 已逐渐被第二代或第三代抗抑郁药物所代替[15]。

|
Figure 1 Metabolic pathway of amitriptyline |
第二代抗抑郁药物为SSRIs, 是目前常用的抗抑郁药物, 主要通过选择性阻断5-羟色胺再摄取发挥抗抑郁作用。临床中常用的SSRIs药物主要有氟西汀、帕罗西汀、舍曲林、西酞普兰等。
氟西汀(fluoxetine)是一种强效SSRIs, 口服后能迅速被吸收并广泛分布于组织中。在CYP2C9酶作用下, 氟西汀通过N-去甲基化反应生成活性代谢物诺氟西汀(norfluoxetine), 进一步均经CYP2C19和CYP3A4的作用发生O-脱烷基化, 生成对三氟甲基苯酚和马尿酸, 随尿液排出[16]。氟西汀的代谢过程见图 2。

|
Figure 2 Metabolic pathway of fluoxetine |
帕罗西汀(paroxetine)也是一种强效SSRIs[17], 具有两个手性中心, 临床中常用(3S, 4R)的异构体[18]。帕罗西汀的亚甲二氧基被取代过程主要由CYP1A2、CYP2C19和CYP3A4催化, 生成儿茶酚类物质, 再通过儿茶酚氨氧位甲基转移酶(COMT)的作用生成甲基化代谢物, 随后经硫酸化或葡萄糖醛酸化, 产物随尿液排出[19], 其代谢过程如图 3所示。其中, 帕罗西汀的亚甲二氧基基团在代谢中可形成卡宾中间体, 与CYP2D6酶发生不可逆结合使酶失活。研究发现, 两个氢被氘代的氘代帕罗西汀(CTP-347), 可在药物不失活性的情况下大大减少卡宾的生成, 降低酶失活毒性, 改善了与其他经CYP2D6代谢药物的联合用药问题[20]。

|
Figure 3 Metabolic pathway of paroxetine |
舍曲林(sertraline)是一种具有两个手性中心的萘胺类SSRIs[21], 构型为cis-(1S, 4S)的舍曲林药效优且安全性高[22]。舍曲林在肝脏中经CYP3A4、CYP2C19催化, 生成去甲基化代谢物, 在MAO作用下被氧化为酮, 随后生成葡萄糖醛酸结合产物随尿液排出[18, 21]。舍曲林的代谢过程示意见图 4。

|
Figure 4 Metabolic pathway of sertraline |
此外, SSRIs药物西酞普兰在肝脏中, 通过CYP2C19和CYP3A4发生N-去甲基化反应, 在CYP2D6作用下进一步发生N-去甲基化, 在单胺氧化酶(MAO)作用下进一步代谢为丙酸类派生物[18], 随后通过肾排泄途径排出[23], 其代谢过程示意图见图 5。

|
Figure 5 Metabolic pathway of citalopram |
第三代抗抑郁药物选择性SNRIs代表性药物为文拉法辛(venlafaxine), 可通过抑制受体对去甲肾上腺素和5-羟色胺的再摄取发挥抗抑郁作用[24]。文拉法辛在肝脏中经CYP2D6生成O-去甲文拉法辛, 在CYP2C19作用下生成N-去甲文拉法辛, 其原型与代谢产物均主要经肾排泄途径(87%)排出体外[25], 代谢过程如图 6。其中, 活性代谢物O-去甲文拉法辛(desvenlafaxine)也能通过抑制5-羟色胺和去甲肾上腺素的再摄取发挥抗抑郁作用, 因其耐受性好, 且有利于与经CYP2D6催化的药物联合用药, 目前已被开发为抗抑郁药物(被美国惠氏公司开发为商品名为Pristiq®的抗抑郁药物)用于临床治疗中[26]。

|
Figure 6 Metabolic pathway of venlafaxine |
与SNRIs通过抑制神经元对5-羟色胺和去甲肾上腺素的再摄取的机制不同, NaSSA则主要通过阻滞5-羟色胺2A (5-HT2A)和α2-肾上腺素受体的再摄取, 提高突触间去甲肾上腺素和5-羟色胺神经递质水平[27], 发挥抗抑郁作用[5, 10]。米氮平(mirtazapine)是一种典型的NaSSA, 能用于治疗伴有焦虑的抑郁症。它在CYP3A4和CYP1A2的作用下, 发生氮氧化反应或N-去甲基作用, 生成氮氧化物或N-去甲基米氮平, 在CYP2D6作用下8位被羟基化, 进一步在葡萄糖醛酸转移酶(UGT)作用下生成葡萄糖醛酸化代谢产物[28], 其代谢过程示意图见图 7。

|
Figure 7 Metabolic pathway of mirtazapine |
安非他酮(bupropion)是一种具有苯乙胺结构的氨基酮类抗抑郁药, 通过抑制多巴胺和去甲肾上腺素的再摄取发挥抗抑郁疗效[29]。安非他酮在肝脏中主要经CYP2B6催化生成羟基化代谢物(少部分被CYP2E1羟基化), 使叔丁基部分甲基被羟基化[30], 代谢过程如图 8所示。研究证实[31], 羟基化代谢物为活性代谢物, 也能阻断5-HT3ARs受体发挥抗抑郁作用。在排泄时, 主要以其代谢产物葡萄糖醛酸结合代谢产物及间氯马脲酸等随尿液和粪便被排出[27]。

|
Figure 8 Metabolic pathway of bupropion |
圣约翰草作为一种天然药物, 在治疗中度抑郁症方面疗效确切[32]。目前临床使用的圣约翰草是贯叶连翘提取物制剂, 主要成分有黄酮类化合物(金丝桃苷、芦丁、异槲皮苷、槲皮苷等)、萘骈二蒽酮类(金丝桃素、伪金丝桃素等)和间苯三酚类衍生物(贯叶金丝桃素)[33]。其中, 间苯三酚类衍生物贯叶金丝桃素(hyperforin)被认为是发挥抗抑郁作用的主要成分之一, 可通过抑制神经元对5-羟色胺、多巴胺及去甲肾上腺素的再摄取发挥抗抑郁疗效[34]。贯叶金丝桃素的代谢过程主要由CYP2C和CYP3A家族参与, 在支链烷基及不饱和键处发生氧化反应[35]。
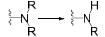
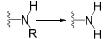
3 抗抑郁药物与CYP450酶的相互作用 3.1 抗抑郁药物的代谢酶CYP450酶广泛存在于机体内, 并参与外源性药物的生物转化[36], 可介导抗抑郁药物的Ⅰ相代谢过程。抗抑郁药物的化学结构中通常含有杂环氮、叔胺或仲胺, 但其整体结构的极性及亲和性各异, 因此相同的代谢途径也可能由不同的CYP450酶介导[37]。表 1总结了部分抗抑郁药物的结构与CYP450酶的关系[21, 25, 28, 38-45], 与文献报道一致[38], 其中CYP2D6、CYP2C19和CYP3A4酶在抗抑郁药物的代谢过程发挥着重要作用, 另外, CYP2C9和CYP2B6等也在部分抗抑郁药物的代谢中发挥着作用。
| Table 1 The properties of CYP450 enzyme and the structure of antidepressants. *Active metabolite. TCAs: Tricyclic antidepressants; SSRIs: Selective serotonin reuptake inhibitors; SNRIs: Serotonin and noradrenaline reuptake inhibitors; NaSSA: Noradrenergic and specific serotonergic antidepressants; NDRIs: Noradrenaline reuptake inhibitors |





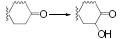
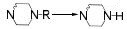


药物的生物转化过程由多种代谢酶催化, 但代谢酶的遗传多态性及药物对代谢酶活性的影响是引起药物药动学特征变异、药物不良反应和治疗失败的主要原因。因此, 明确代谢酶的多态性及药物与酶的相互作用对指导临床个体化用药具有积极的作用[46]。
3.2 CYP450酶的遗传多态性与个体化用药人体内的药物代谢酶具有广泛的遗传多态性, 其代谢药物的能力因个体及种族存在明显差异, 可分为超常代谢型(ultra-rapid metabolizers, UM)、正常代谢型(extensive metabolizers, EM)、中间代谢型(interme-diate metabolizers, IM)和慢代谢型(poor metabolizers, PM)[47]。药物代谢酶的遗传多态性是用药个体差异的分子基础, 明确酶的个体间差异, 利于减少不良反应并指导临床合理用药[48]。
CYP2D6仅占肝脏代谢酶的1%~2%, 但经其代谢的药物却多达25%, 具有广泛的多态性, 且不同个体间活性相差可达1 000倍[49], 目前已证实其有105不同的等位基因, 约有1%亚洲人是CYP2D6慢代谢型患者[50]。临床用药中也发现了服用相同剂量抗抑郁药物在CYP2D6正常代谢者和慢代谢者体内表现出显著性差异, 如:多数CYP2D6慢代谢型抑郁症患者服用文拉法辛后出现了反胃、恶心、呕吐等不良反应症状[51]; 临床中也曾出现CYP2D6慢代谢型抑郁症患者服用氟西汀后因体内原型及活性代谢物诺氟西汀浓度比预期超出数倍而发生致命现象[52]。
约10%常用药物的生物转化由CYP2C19介导, 它具有广泛的遗传多态性, 且在不同种族间差异显著, 目前已证实其有30余种遗传突变体[53], 约有30%的亚洲人携带CYP2C19突变基因(CYP2C19*2和CYP2C19*3基因型通常表现为慢代谢型)[37]。CYP2C19在抗抑郁药物代谢中主要介导药物的去烷基化反应, 研究已证实[54], CYP2C19慢代谢型抑郁症患者服用抗抑郁药物后产生不良反应的现象明显高于正常代谢者。当CYP2C19慢代谢型抑郁症患者服用氟西汀后, 其半衰期和AUC较正常代谢型患者显著增高, 清除率降低约50%;这种药动学参数变化趋势也同样在服用西酞普兰的CYP2C19慢代谢型抑郁症患者中有表现[49, 55]。
CYP3A4主要在人体肝脏与肠道中表达, 在肝脏中约占30%, 在肠道中约占70%, 参与临床约50%药物的代谢, 在抗抑郁药物的生物转化中发挥了重要作用。CYP3A4的表达在个体间的差异约20~40倍[56], 慢代谢型在中国人中占比为3.4%, 且具有种族差异, 因此, 以中国人为对象的临床实验具有非常重要的意义[57]。
CYP2C9是CYP2C家族的主要成员, 约占肝中CYP450总量的20%, 催化约12%的临床常用药物, 中国人中CYP2C9慢代谢型的发生率(CYP2C9*3)大约有3%~5%[58]。研究显示[59], 抗抑郁药物氟西汀的生物转化除受CYP2D6和CYP2C19多态性的影响外, 也受CYP2C9遗传多态性影响。
CYP2B6约参与7%临床常用药物的代谢, 具有高度基因多态性, 且有明显的种族和地域差异[60]。相关研究调查显示[45], CYP2B6*6和CYP2B6*18基因型能影响安非他酮羟基化代谢物的血药稳态浓度, 使其稳态浓度约降低33%。
由此可见, CYP450酶的多态性是引起药物个体差异的重要因素。因此, 将代谢酶的多态性与临床用药相结合, 根据患者代谢酶的基因型选择药物, 从而预测药物作用的强度, 调整用药剂量, 提高临床用药的安全性和可靠性。
3.3 抗抑郁药物对CYP450酶的影响抑郁症发病的同时常伴随其他疾病发生。因此, 在抑郁症的治疗中, 抗抑郁药物通常会结合其他药物共同治疗机体系统的紊乱, 因联合用药诱发的药物间潜在的相互作用也时有发生, 从而影响药物的有效性和安全性。因此, 在临床用药中, 研究抗抑郁药物对代谢酶活性的影响不可忽视。
抗抑郁药物多引起CYP2D6酶的抑制作用与CYP3A4的抑制或诱导作用, 从而影响合并用药的药动学过程, 引起不良反应。抗抑郁药物与治疗精神分裂症药物合用时发现, 氟西汀及其代谢产物诺氟西汀能显著抑制CYP2D6酶的活性, 对CYP2C9酶具有中等程度的抑制作用, 而对CYP2C19和CYP3A4酶抑制作用较弱, 当与经CYP2D6代谢的治疗精神分裂症药物氟哌啶醇、氯氮平或利培酮合用时, 导致其代谢抑制[61]。在阿托西汀和氟西汀联合用药时, 氟西汀会通过抑制CYP2D6的活性影响阿托西汀原型及其代谢物4-羟基阿托西汀葡萄糖醛酸结合物代谢, 使AUC增加10%~30%[62]。此外, SSRIs帕罗西汀与治疗精神分裂症药物奋乃静合用时, 因帕罗西汀抑制CYP2D6的活性, 导致奋乃静血药浓度升高2~13倍, 使患者出现锥体外系反应[63]。氟伏沙明对CYP2D6和CYP3A4也具有抑制作用, 与治疗精神分裂症药物氯氮平合用时会使其血药浓度升高, 并出现恶心头晕等不良反应[64]; 与喹硫平合用时, 使之血药浓度升高159%[65]; 与抗焦虑药物坦度螺酮合用时, 导致CYP3A4介导的生物转化减少, 血药浓度升高[66]。
相关学者在临床研究中发现, 肿瘤患者也常患有抑郁症。治疗乳腺癌药物他莫昔芬活性代谢物4-羟基-N-去甲基他莫昔芬的生成主要由CYP2D6参与, 当与抗抑郁药物帕罗西汀合用时, 会导致他莫昔芬活性代谢产物的浓度显著降低[67]。另据相关报道[68], 抗癌药物多比柔星与抗抑郁药物安非他酮会相互影响对方代谢过程, 多比柔星对CYP2B6的抑制作用影响了安非他酮的代谢过程, 而安非他酮对CYP2D6抑制也影响着多比柔星的代谢。
此外, 治疗心血管药物地高辛、阿托伐他丁等的临床疗效, 也因SSRIs对CYP2D6和CYP3A4有抑制作用而受到影响[69, 70]。
另有研究表明, 健康志愿者连续2周服用天然抗抑郁药物圣约翰草, 对CYP3A4有显著的诱导作用, 且P-gp的外排也受到了诱导[71]。与免疫抑制剂药物他克莫司合用时, 引起他克莫司口服生物利用度降低、清除率加快与血药浓度的降低[3]。
4 结语研究抗抑郁药物的体内代谢是药物开发及临床安全用药必不可少的过程。许多活性化合物在体外试验中显示具有高活性, 但因其体内代谢特征欠佳或因产生毒性代谢物而失去开发价值。因此, 在研发前期通过研究抗抑郁药物的体内代谢、药物代谢途径、相关代谢酶及代谢过程中产生的活性代谢物, 明确原形药物可能的代谢位点, 降低毒性或增加代谢稳定性, 可提高化合物开发的成活率; 另一方面, 在研究过程中发现的活性代谢物, 也可作为先导化合物进一步进行药理毒理学等相关研究, 将有助于发现药动学过程更好的活性化合物或了解前体药物的体内过程, 以明确药物的作用机制。通过上述研究, 明确药物的代谢过程、药物代谢过程中的关键酶及其多态性与药理毒理学特征, 最终阐明其在体内的作用靶点与通路, 为药物作用机制的阐明及新型抗抑郁药物的发现提供思路。除此之外, 对药物与药物、药物与酶的相互作用等方面的评估也必不可少, 可为临床用药种类与剂量的选择及合理安全用药等提供有力依据。
| [1] | Lam RW, Malhi GS, McIntyre RS, et al. Fatigue and occupational functioning in major depressive disorder[J]. Aust N Z J Psychiatry, 2013, 47: 989–991. DOI:10.1177/0004867413488222 |
| [2] | Ma LN, Li Yun. The progress of antidepressant drug[J]. Med Recapitul (医学综述), 2011, 17: 3777–3779. |
| [3] | Zeng S. Drug Metabolism (药物代谢学)[M]. Hangzhou: Zhejiang University Press, 2008: 6-250. |
| [4] | Shah MM. HCN1 Channels:a new therapeutic target for depressive disorders[J]. Sci Signal, 2012, 244: 44. |
| [5] | Fajemiroye JO, Silva DM, Oliveira DR, et al. Treatment of anxiety and depression:medicinal plants in retrospect[J]. Fundam Clin Pharmacol, 2016, 30: 198–215. DOI:10.1111/fcp.2016.30.issue-3 |
| [6] | Jia HM, Feng YF, Liu YT, et al. Integration of 1H NMR and UPLC-Q-TOF/MS for a comprehensive urinary metabonomics study on a rat model of depression induced by chronic unpredictable mild stress[J]. PLoS One, 2013, 8: e63624. DOI:10.1371/journal.pone.0063624 |
| [7] | Schmidt M, Butterweck V. The mechanisms of action of St. John's wort:an update[J]. Wien Med Wochenschr, 2015, 165: 229–235. DOI:10.1007/s10354-015-0372-7 |
| [8] | Zhou JQ, Wang C. Clinical observation of remifemin combined with paroxetine for perimenopausal syndrome complicating with depression[J]. China Pharm (中国药房), 2013, 24: 3419–3421. |
| [9] | Fu JH. Study on the Mechanism of a New Type Antidepressant Shuganjiuyu Capsule in a Rat Model of Depression (新型抗抑郁中药舒肝解郁胶囊对抑郁模型大鼠的作用机制研究)[D]. Changsha: Central South University, 2014: 44-45. |
| [10] | Richelson E. Pharmacology of antidepressants[J]. Mayo Clin Proc, 2001, 76: 511–527. DOI:10.4065/76.5.511 |
| [11] | Kallem RR, Jillel AB, Ravula AR, et al. Highly sensitive LC-MS/MS-ESI method for determination of phenelzine in human plasma and its application to a human pharmacokinetic study[J]. J Chromatogr B Analyt Technol Biomed Life Sci, 2016, 1022: 126–132. DOI:10.1016/j.jchromb.2016.04.006 |
| [12] | Breyer Pfaff U. The metabolic fate of amitriptyline, nortriptyline and amitriptylinoxide in man[J]. Drug Metab Rev, 2004, 36: 723–746. DOI:10.1081/DMR-200033482 |
| [13] | Steimer W, Zopf K, Amelunxen S, et al. Allele-specific change of concentration and functional gene dose for the prediction of steady-state serum concentrations of amitriptyline and nortriptyline in CYP2C19 and CYP2D6 extensive and intermediate metabolizers[J]. Clin Chem, 2004, 50: 1623–1633. DOI:10.1373/clinchem.2003.030825 |
| [14] | Zhou X, Chen C, Zhang FR, et al. Metabolism and bioactivetion of the tricyclic antidepressant amitriptyline in human liver microsomes and human urine[J]. Bioanal, 2016, 8: 1365–1381. DOI:10.4155/bio-2016-0025 |
| [15] | Soara J, Perkinsb GD, Abbasc G, et al. European Resuscitation Council guidelines for resuscitation 2010. Section 8. Cardiac arrest in special circumstances[J]. Resuscitation, 2010, 81: 1400–1433. DOI:10.1016/j.resuscitation.2010.08.015 |
| [16] | Margolis JM, Donnell JP, Mankowski DC, et al. R -, S -, and racemic fluoxetinen-demethylation by human cytochrome p450 enzymes[J]. Drug Metab Dispos, 2000, 28: 1187–1191. |
| [17] | Pae CU, Patkar AA. Paroxetine:current status in psychiatry[J]. Expert Rev Neurother, 2007, 7: 107–120. DOI:10.1586/14737175.7.2.107 |
| [18] | Hiemke C, H rtter S. Pharmacokinetics of selective serotonin reuptake inhibitors[J]. Pharmacol Ther, 2000, 85: 11–28. DOI:10.1016/S0163-7258(99)00048-0 |
| [19] | Jornil J, Jensen KG, Larsen F, et al. Identification of cytochrome P450 isoforms involved in the metabolism of paroxetine and estimation of their importance for human paroxetine metabolism using a population-based simulator[J]. Drug Metab Dispos, 2010, 38: 376–385. DOI:10.1124/dmd.109.030551 |
| [20] | Jiang WF, Li WB. Application of deuteration in drug research[J]. Qilu Pharma Aff (齐鲁药事), 2010, 29: 682–684. |
| [21] | Mandrioli R, Mercolini L, Raggi MA. Evaluation of the pharmacokinetics, safety and clinical efficacy of sertraline used to treat social anxiety[J]. Expert Opin Drug Metab Toxicol, 2013, 9: 1495–1505. DOI:10.1517/17425255.2013.816675 |
| [22] | Rao RN, Talluri MV, Maurya PK. Separation of stereoisomers of sertraline and its related enantiomeric impurities on a dimethylated beta-cyclodextrin stationary phase by HPLC[J]. J Pharm Biomed Anal, 2009, 50: 281–286. DOI:10.1016/j.jpba.2009.04.038 |
| [23] | Mrazek DA, Biernacka JM, Kane DJ, et al. CYP2C19 variation and citalopram response[J]. Pharmacogenet Genom, 2011, 21: 1–9. DOI:10.1097/FPC.0b013e328340bc5a |
| [24] | Korte SM, Prins J, Krajnc AM, et al. The many different faces of major depression:it is time for personalized medicine[J]. Eur J Pharmacol, 2015, 753: 88–104. DOI:10.1016/j.ejphar.2014.11.045 |
| [25] | Karlsson L, Zackrisson AL, Josefsson M, et al. Influence of CYP2D6 and CYP2C19 genotypes on venlafaxine metabolic ratios and stereoselective metabolism in forensic autopsy cases[J]. Pharmacogenom J, 2015, 15: 165–171. DOI:10.1038/tpj.2014.50 |
| [26] | Cooper JM, Brown JA, Cairns R, et al. Desvenlafaxine overdose and the occurrence of serotonin toxicity, seizures and cardiovascular effects[J]. Clin Toxicol (Phila), 2016, 53: 18–24. |
| [27] | Wille SM, Cooreman SG, Neels HM, et al. Relevant issues in the monitoring and the toxicology of antidepressants[J]. Crit Rev Clin Lab Sci, 2008, 45: 25–89. DOI:10.1080/10408360701713112 |
| [28] | Jaquenoud SE, Harenberg S, Vandel P, et al. Multicenter study on the clinical effectiveness, pharmacokinetics, and pharmacogenetics of mirtazapine in depression[J]. J Clin Psychopharmacol, 2012, 32: 622–629. DOI:10.1097/JCP.0b013e3182664d98 |
| [29] | Stahl SM, Pradko JF, Haight BR, et al. A review of the neuropharmacology of bupropion, a dual norepinephrine and dopamine reuptake inhibitor[J]. Prim Care Companion J Clin Psych, 2004, 6: 159–166. DOI:10.4088/PCC.v06n0403 |
| [30] | Hesse LM, Venkatakrishnan K, Court MH, et al. CYP2B6 mediates the in vitro hydroxylation of bupropion:potential drug interactions with other antidepressants[J]. Drug Metab Dispos, 2000, 28: 1176–1183. |
| [31] | Pandhare A, Pappu AS, Wilms H, et al. The antidepressant bupropion is a negative allosteric modulator of serotonin type 3A receptors[J]. Neuropharmacol, 2016, 113: 89–99. |
| [32] | Oliveira AI, Pinho C, Sarmento B, et al. Neuroprotective activity of hypericum perforatum and its major components[J]. Front Plant Sci, 2016, 7: 1–15. |
| [33] | Chuan YY, Li Y. Research review of Hypericum perforatum (St. John's Wort)[J]. J Northwest Pharm (西北药学杂志), 2016, 31: 330–330. |
| [34] | Sarris J. St. John's wort for the treatment of psychiatric disorders[J]. Psychiatr Clin North Am, 2013, 36: 65–72. DOI:10.1016/j.psc.2013.01.004 |
| [35] | Hokkanen J, Tolonen A, Mattila S, et al. Metabolism of hyperforin, the active constituent of St. John's wort, in human liver microsomes[J]. Eur J Pharm Sci, 2011, 42: 273–284. DOI:10.1016/j.ejps.2010.12.002 |
| [36] | Zhou L, Zhong DF, Chen XY. Research advances in non-P450-mediated drug oxidative metabolism[J]. Acta Pharm Sin (药学学报), 2017, 52: 8–18. |
| [37] | Oliveira AI, Pinho C, Sarmento B, et al. Neuroprotective activity of Hypericum perforatum and its major components[J]. Front Plant Sci, 2016, 7: 1–15. |
| [38] | Zanger UM, Turpeinen M, Klein K, et al. Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation[J]. Anal Bioanal Chem, 2008, 392: 1093–1108. DOI:10.1007/s00216-008-2291-6 |
| [39] | Hicks JK, Bishop JR, Sangkuhl K, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors[J]. Clin Pharmacol Ther, 2015, 98: 127–134. DOI:10.1002/cpt.v98.2 |
| [40] | Bertilsson L. Metabolism of antidepressant and neuroleptic drugs by cytochrome p450s:clinical and interethnic aspects[J]. Clin Pharmacol Ther, 2007, 82: 606–609. DOI:10.1038/sj.clpt.6100358 |
| [41] | Hicks JK, Swen JJ, Thorn CF, et al. Clinical pharmacogenetics implementation consortium guideline for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants[J]. Clin Pharmacol Ther, 2013, 93: 402–408. DOI:10.1038/clpt.2013.2 |
| [42] | Pelkonen O, Turpeinen M, Hakkola J, et al. Inhibition and induction of human cytochrome P450 enzymes:current status[J]. Arch Toxicol, 2008, 82: 667–715. DOI:10.1007/s00204-008-0332-8 |
| [43] | Lobo ED, Bergstrom RF, Reddy S, et al. In vitro and in vivo evaluations of cytochrome P4501A2 interactions with duloxetine[J]. Clin Pharmacokinet, 2008, 47: 191–202. DOI:10.2165/00003088-200847030-00005 |
| [44] | Turpeinen M, Raunio H, Pelkonen O. The functional role of CYP2B6 in human drug metabolism:substrates and inhibitors in vitro, in vivo and in silico[J]. Curr Drug Metab, 2006, 7: 705–714. DOI:10.2174/138920006778520633 |
| [45] | Spina E, Leon J. Clinical applications of CYP genotyping in psychiatry[J]. J Neural Transm (Vienna), 2015, 122: 5–28. DOI:10.1007/s00702-014-1300-5 |
| [46] | Weinshilboum R. Inheritance and drug response[J]. N Engl J Med, 2003, 348: 529–537. DOI:10.1056/NEJMra020021 |
| [47] | Cheng D, Xu WR, Liu CX. Progress of research on genetic polymorphism of CYP450s[J]. Chin Pharmacol Bull (中国药理学通报), 2006, 22: 1409–1414. |
| [48] | Ota T, Kamada Y, Hayashida M, et al. Combination analysis in genetic polymorphisms of drug-metabolizing enzymes CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A5 in the Japanese population[J]. Int J Med Sci, 2015, 12: 78–82. DOI:10.7150/ijms.10263 |
| [49] | Yan F, Xian CH, Xiong YQ. Effect of CYP2C19 genetic polymorphism on drug metabolism and individualized therapy[J]. Chin J Clin Pharmacol Ther (中国临床药理学与治疗学), 2010, 15: 949–953. |
| [50] | Haufroid V, Hantson P. CYP2D6 genetic polymorphisms and their relevance for poisoning due to amphetamines, opioid analgesics and antidepressants[J]. Clin Toxicol (Phila), 2015, 53: 501–510. DOI:10.3109/15563650.2015.1049355 |
| [51] | Shams ME, Arneth B, Hiemke C, et al. CYP2D6 polymorphism and clinical effect of the antidepressant venlafaxine[J]. J Clin Pharm Ther, 2006, 31: 493–502. DOI:10.1111/jcp.2006.31.issue-5 |
| [52] | Sallee FR, Devane CL, Ferrell RE. Fluoxetine-related death in a child with cytochrome P-4502D6 genetic deficiency[J]. J Child Adolesc Psychopharmacol, 2000, 10: 27–34. DOI:10.1089/cap.2000.10.27 |
| [53] | Dai DP, Hu LM, Geng PW, et al. In vitro functional analysis of 24 novel CYP2C19 variants recently found in the Chinese Han population[J]. Xenobiotica, 2015, 45: 1030–1035. DOI:10.3109/00498254.2015.1028512 |
| [54] | Zhou Q, Yu LS, Zeng S. Personalized dosing from perspective of pharmacogenomics of drug metabolizing enzymes and transporters[J]. Acta Pharm Sin (药学学报), 2017, 52: 1–7. |
| [55] | Desta Z, Zhao X, Shin JG, et al. Clinical significance of the cytochrome P4502C19 genetic polymorphism[J]. Clin Pharmacokinet, 2002, 41: 913–958. DOI:10.2165/00003088-200241120-00002 |
| [56] | Dong ZL, Wang GX, Fang XM. Progress in research of cytochrome P4503A4 gene polymorphism[J]. Int J Anesth Resusc (国际麻醉学与复苏杂志), 2012, 33: 705–709. |
| [57] | Chen X, Li YZ, Fang Y. The characteristics and polymorphism of CYP3A4 for the metabolic drugs[J]. China Pharm (中国药房), 2010, 22: 2097–2099. |
| [58] | Kim SH, Kim DH, Byeon JY, et al. Effects of CYP2C9, genetic polymorphisms on the pharmacokinetics of celecoxib and its carboxylic acid metabolite[J]. Arch Pharm Res, 2016, 40: 382–390. |
| [59] | LLerena A, Dorado P, Berecz R, et al. Effect of, CYP2D6, and, CYP2C9, genotypes on fluoxetine and norfluoxetine plasma concentrations during steady-state conditions[J]. Eur J Clin Pharmacol, 2004, 59: 869–871. DOI:10.1007/s00228-003-0707-y |
| [60] | Qin WJ, Zhou HH. Advances in study of CYP2B6 gene polymorphisms and its functional significances[J]. Chin J Clin Pharmacol Ther (中国临床药理学与治疗学), 2008, 13: 1434–1440. |
| [61] | Zhang SN, Yao H, Li JM, et al. Drug interaction during combing fluoxetine and risperidone to treat schizophrenia[J]. Chin J Clin Pharmacol (中国临床药理学杂志), 2009, 25: 11–13. |
| [62] | Todor I, Popa A, Neag M, et al. Evaluation of the potential pharmacokinetic interaction between atomoxetine and fluvoxamine in healthy volunteers[J]. Pharmacology, 2016, 99: 84–88. |
| [63] | Micallef J, Fakra E, Blin O. Use of antidepressant drugs in schizophrenic patients with depression[J]. Encephale, 2006, 32: 263–269. DOI:10.1016/S0013-7006(06)76153-X |
| [64] | Lin H, Lu SP, Shang XF, et al. The interaction of clozapine combined with fluvoxamine in the treatment of schizophrenia[J]. Chin Pharm J (中国药学杂志), 2008, 43: 1919–1920. |
| [65] | Spina E, De LJ. Clinically relevant interactions between newer antidepressants and second-generation antipsychotics[J]. Expert Opin Drug Metab Toxicol, 2014, 10: 721–746. DOI:10.1517/17425255.2014.885504 |
| [66] | Nishikawa H, Inoue T, Masui T, et al. Pharmacokinetic interaction between tandospirone and fluvoxamine in the rat contextual conditioned fear stress model and its functional consequence:involvement of cytochrome P4503A4[J]. Psychiat Clin Neurosci, 2008, 62: 591–596. DOI:10.1111/j.1440-1819.2008.01853.x |
| [67] | Juurlink D. Revisiting the drug interaction between tamoxifen and SSRI antidepressants[J]. Br Med J, 2016, 354: 5309–5310. |
| [68] | He J, Yu Y, Prasad B, et al. Mechanism of an unusual, but clinically significant, digoxin-bupropion drug interaction[J]. Biopharm Drug Dispos, 2014, 35: 253–263. DOI:10.1002/bdd.v35.5 |
| [69] | Sager JE, Tripathy S, Price L S, et al. In vitro to in vivo extrapolation of the complex drug-drug interaction of bupropion and its metabolites with CYP2D6; simultaneous reversible inhibition and CYP2D6 downregulation[J]. Biochem Pharmacol, 2016, 123: 85–96. DOI:10.1016/j.biochi.2016.02.002 |
| [70] | Herbet M, Gawrońska-Grzywacz M, Izdebska M, et al. Effect of the interaction between atorvastatin and selective serotonin reuptake inhibitors on the blood redox equilibrium[J]. Exp Ther Med, 2016, 12: 3440–3444. DOI:10.3892/etm.2016.3794 |
| [71] | Lucire Y, Crotty C. Antidepressant-induced akathisia-related homicides associated with diminishing mutations in metabolizing genes of the CYP450 family[J]. Pharmgenom Pers Med, 2011, 4: 65–81. |