 2017, Vol. 52
2017, Vol. 52

编者按
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
Anacor公司从生化机制出发研究抗菌药, 后转向抗真菌药收获了抗甲癣药他伐硼罗; 还从炎症的靶标磷酸二酯酶4和炎症因子入手, 研发出治疗过敏性皮炎的克立硼罗。研发过程探索了多种可能与渠道, 虽然有些与最终目标未能搭界, 但始终坚持研究有机硼酸酯类的执着精神, 积累了许多经验, 终于诞生两个首创药。研发过程涉及的药物化学、分子生物学和作用机制等研究思路, 值得借鉴。由于克立硼罗的安全有效和潜在的市场价值, 辉瑞公司以52亿美元收购了该公司。本文以药物化学视角简析两个药物的创制过程。
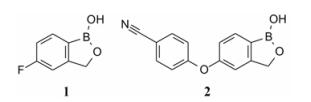
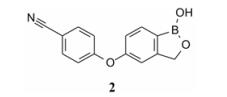
他伐硼罗(1, tavaborole)和克立硼罗(2, crisaborole)可视作两个姊妹药, 分别于2014和2016年经美国FDA批准为外用的新分子实体, 分别治疗甲癣(灰趾/指甲)和过敏性皮炎(湿疹)。两个药物都是首创的有机硼酸酯, 结构骨架相似, 出自一个公司Anacor。甲癣和湿疹虽不危及生命, 却是常见的复发性慢性皮肤疾患。作为含硼的有机小分子, 相对分子质量为251以内, 在近年来众多上市抗肿瘤和代谢性疾病等药物中, 其化学结构和适应证颇显“另类”。但从药物化学的视角却值得借鉴。
|
项目起始于研究抗菌药, 是从细菌生长机制入手。新月柄杆菌(Caulobacter crescentus)类致病菌的甲基转移酶(CcrM)是催化细菌DNA序列中GANTC的腺嘌呤N6发生甲基化的酶系, 甲基化的DNA对维持细菌增殖和毒力起重要作用, 因而CcrM是研制抗菌药的一个靶标。CcrM酶的催化机制是与底物DNA和辅酶S-腺苷蛋氨酸(AdoMet)形成三元复合物, 将甲基从AdoMet分子上转移到DNA的腺嘌呤N6 (图 1)。

|
图 1 CcrM催化转甲基的反应过程 |
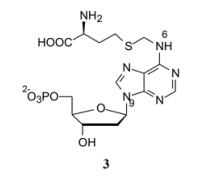
由于缺乏CcrM三维结构信息, 研制者设想在DNA和辅酶AdoMet的结合部位有一个超螺旋腺嘌呤结合位点, 因而设计了拼合型的CcrM酶的多底物抑制分子, 将辅酶中的甲硫氨酸经亚甲基连接到腺嘌呤环的N6上, 设计了模拟辅酶结构的假底物3, 实验表明是CcrM选择性抑制剂, 而对胞苷甲基转移酶(Hha1) 没有抑制活性, 遂将3作为苗头化合物展开研究(Wahnon DC, Shier VK, Benkovic SJ. Mechanism-based inhibition of an essential bacterial adenine DNA methyltransferase: rationally designed antibiotics. J Am Chem Soc, 2001, 123: 976−977)。
|
苗头物3包含底物和辅酶共有的腺嘌呤片段。在3转化为先导物(hit-to-lead)的操作中, 以腺嘌呤为骨架变换N6和N9连接的基团, 连接不同体积和形状的侧链, 合成了1 000多个结构多样的化合物库, 进行活性评价。
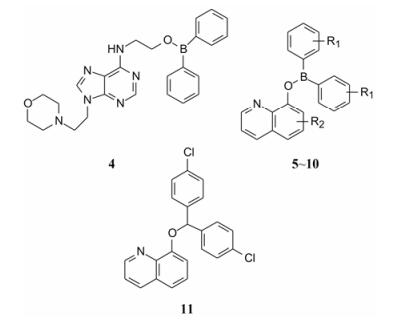
评价活性采用两种模型:对新月柄杆菌生长的抑制和对CcrM酶活性的抑制。初筛受试物浓度为100 μmol·L−1, 测定对细菌和酶的抑制率。发现了6个活性较高的化合物, 却都是含有二苯基硼酯的结构, 例如化合物4。以4为新的苗头, 设计以二苯基硼酸-8-羟基喹啉酯为母核的化合物, 其中有代表性的化合物如5~11 (表 1), 通过研究构效关系优化活性。
| 表 1 受试物(浓度为100 μmol·L−1)对3种甲基转移酶的抑制活性。*细菌的腺嘌呤甲基转移酶; **细菌的胞苷甲基转移酶 |
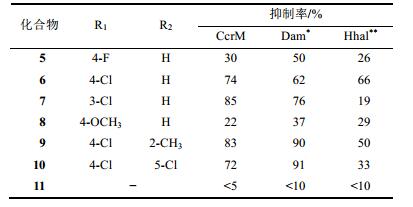
表 1的构效关系表明, 苯环上用氯原子取代(化合物6、7、9和10)对CcrM抑制率超过50%, 其中7、9和10对Dam也有抑制活性, 但对Hhal的作用较弱。化合物11是不含硼的6的同型物, 未显示活性, 提示硼元素的重要性。
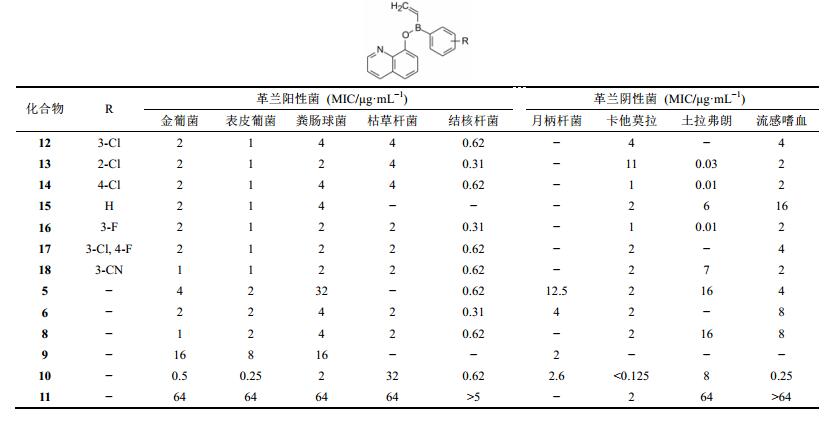
2.3 简化结构为了考察与硼相连的基团大小对抑菌活性的影响, 将一个苯环变换为乙烯基, 合成的化合物连同表 1的分子评价对多种细菌的活性, 表 2列出了化合物的最低抑菌浓度(MIC)。
| 表 2 受试物对细菌的最低抑菌浓度 |
|
表 2中化合物对革兰阳性菌和阴性菌的抑制活性, 显示多数对革兰阳性菌的最低抑菌浓度(MIC)在μg·mL−1范围。由于革兰阳性菌不存在CcrM和Dam酶, 一些化合物特别是10, 对多株阳性菌也呈现活性, 意味着是阻断其他环节所致。10和13对革兰阴性菌土拉菌有抑制作用, 提示化合物的结构变换引起抗菌谱的改变, 并非单纯地抑制CcrM。不过, 不含硼原子的化合物11, 对所有菌株仍没有抑制作用。
2.4 探索抑制革兰阳性菌的作用机制为了解释抑制革兰阳性菌的作用机制, 以枯草杆菌为研究对象, 将甲基转移酶编码基因menH、trmD、trmU、cspR、ydiO和ydiP用分子生物学方法分别构建到细胞中, 当甲基转移酶受到抑制, 生物标志物β-D-硫代半乳糖异丙苷(IPTG)产生量减少, 依此作为评价活性的指标。结果表明menH表达的甲基萘醌甲基转移酶(MenH)可被上述硼化物抑制, 过程如图 2所示。

|
图 2 甲基萘醌甲基转移酶的催化和抑制示意图 |
多数革兰阳性菌含有甲基萘醌甲基转移酶(MenH), MenH结合于膜蛋白上, 在呼吸链和光合作用的电子传递中起重要作用, 但哺乳动物细胞没有MenH, 所以是抗阳性致病菌的一个特异性靶标。
用辅酶[3H]-AdoMet和MenH酶测定上述化合物, 显示6、7和12在100 μmol·L−1浓度下抑制MenH酶(革兰阳性菌)活性分别为15%、30%和50%, 同样浓度下6和7对CcrM酶(革兰阴性菌)的抑制率为74%和85%, 说明不同的化合物对甲基转移酶的特异性不同。至此说明苯硼酸酯的抑菌作用可能与对多种甲基转移酶的抑制有关(Benkovic SJ, Baker SJ, Alley MRK, et al. Identification of borinic esters as inhibitors of bacterial cell growth and bacterial methyl transferases, CcrM and MenH. J Med Chem, 2005, 48: 7468−7476)。
3 骨架迁越—喹啉环的剖裂分析苯硼酸喹啉酯的结构, 由于硼原子外层电子含有空轨道, 可与8-羟基喹啉的氧孤电子对发生配位结合, 形成五元环氮硼内酯。如果这是构成活性的主要片段, 设想可将并合的苯环简化为羰基, 从而喹啉环简化为α-吡啶甲酸, 而且在吡啶的3位引入羟基, 可与2位羰基形成分子内氢键, 以模拟喹啉环的平面结构, 这种骨架迁越可用图 3表示。

|
图 3 喹啉环的骨架迁越 |
优化过程仍以抗菌活性为指标, 用试错方式(trial and error)逐步优化, 表 3列出了化合物结构和活性, 简析构效关系如下: ① 鉴于前述的3-(或4-)氯苯基的活性较高, 合成了3, 3'-二氯(19)和4, 4'=二氯(20)化合物。19虽然对其他阳性菌不如20, 但对金葡菌显示很高的活性。② 将一个苯环换成杂环, 如吡啶(21、22)、2-氯吡啶(23)和噻吩环(24), 抗菌活性都显著下降。③ R2简化为甲基化合物(26)失去活性。④ 氯代苯环上再做甲基取代, 化合物25对革兰阳性菌的活性显著提高。变换成其他基团或增加脂溶性或变更氯或甲基的位置(27~35)活性都不如25。25的抗菌谱与活性强度接近红霉素, 因而成为优良的先导化合物。
| 表 3 五元环氮硼内酯系列的抗菌活性 |
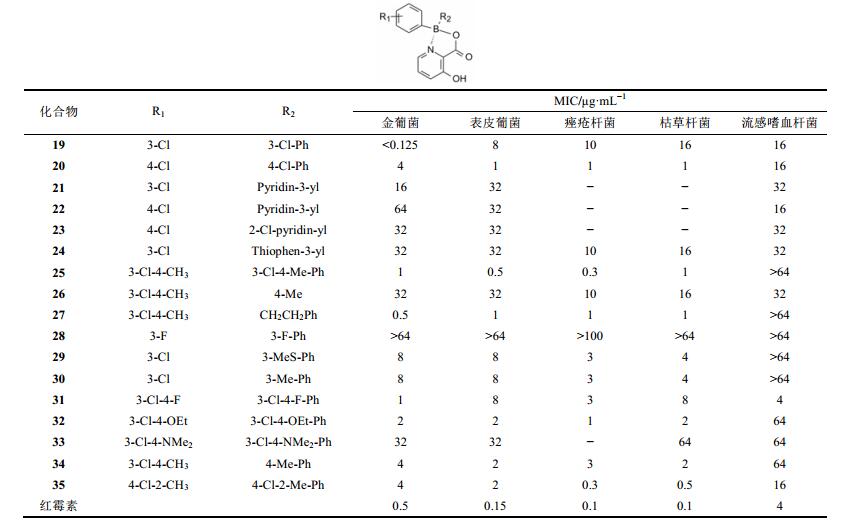
为了优化吡啶环上的取代基, 将两个3-氯-4-甲基苯环固定不变, 探索吡啶环上不同取代基对活性的影响, 合成的化合物及其活性列于表 4。结果表明, 去羟基化合物36, 除保持抑制金葡菌作用外对其他菌株都失去活性。3-乙酰氧基(37)虽然没有氢键供体, 活性仍与25相近, 说明形成六员环的分子内氢键不是活性的必需因素。加大3位取代基体积的苯甲酰化合物38活性显著降低。3-氨基化合物39失去抑制球菌的活性。3-羧基(40)对金葡菌和流感杆菌的抑制活性强于25, 而且羧基移至4-或5-位活性变化不大。综上, 3-羟基化合物25对于皮肤感染的重要致病菌金葡菌和痤疮杆菌活性最高, 成为里程碑式的化合物。
| 表 4 吡啶环上取代基对抗菌活性的影响 |
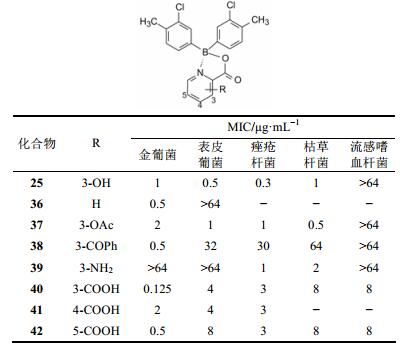
基于以上研究, 将含硼化合物目标定于研发过敏性皮炎(湿疹), 湿疹既是一类炎症, 也大都发生细菌性感染。炎症的发生是由于炎症因子在皮肤上聚集并激活1型或2型辅助性T淋巴细胞(Th1、Th2) 所致, 为此, 评价含硼化合物对细胞因子的抑制作用。
4.1 抗炎活性评价评价化合物的抗炎活性是用抑制外周血单核细胞(PBMC)产生细胞因子的活性作为指标。将PBMC、受试物与炎症诱导剂脂多糖(LPS)和伴刀豆凝集素A或植物血球凝集素(PHA)温孵, 用ELISA方法分别测定TNF-α (Th1细胞因子)或IL-1β、IFN-γ和IL-4 (Th2细胞因子)的释放量, 与空白对照的比值作为化合物的活性值。
4.2 化合物的抗炎活性选取抗菌作用强的化合物, 以10 μmol·L−1浓度测定抑制细胞因子的活性, 结果列于表 5。
| 表 5 代表性化合物的抑制细胞因子活性 |
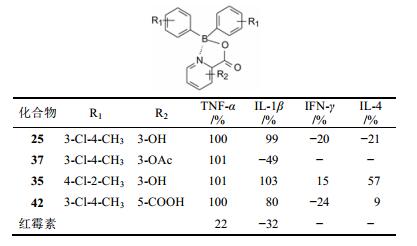
化合物25、35和42抑制促炎性细胞因子TNF-α和IL-1β, 但不抑制IFN-γ和IL-4的释放。37不抑制IL-1β释放, 所以未测对IFN-γ和IL-4的活性。由于25既有抗炎活性, 抗菌作用也强于其他化合物, 拟作为候选化合物进入临床前研究(Baker SJ, Akama T, Zhang YK, et al. Identification of a novel boron-containing antibacterial agent (AN0128) with anti-inflammatory activity, for the potential treatment of cutaneous diseases. Bioorg Med Chem Lett, 2006, 16: 5963−6967)。
4.3 再次骨架迁越化合物25可认为是α-吡啶甲酸与二苯基硼酸的混合酸酐, 并形成N→B配位的环状结构, 研制者为简化结构, 将羰基用亚甲基替换、氮原子用sp2杂化碳等排置换(吡啶换成苯环)并以C-B共价键替换N→B配位键, 去掉一个苯环以满足三价硼的结合, 得到式43的骨架结构。
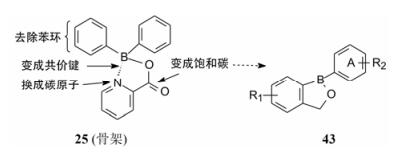
|
通式43的苯环A可用杂环、插烯或乙烯基替换, 也可变换R1和R2基团, 以期在多样性变换中获得优化。合成的化合物列于表 6中。
| 表 6 苯并硼氧环戊烷化合物的抗真菌活性 |
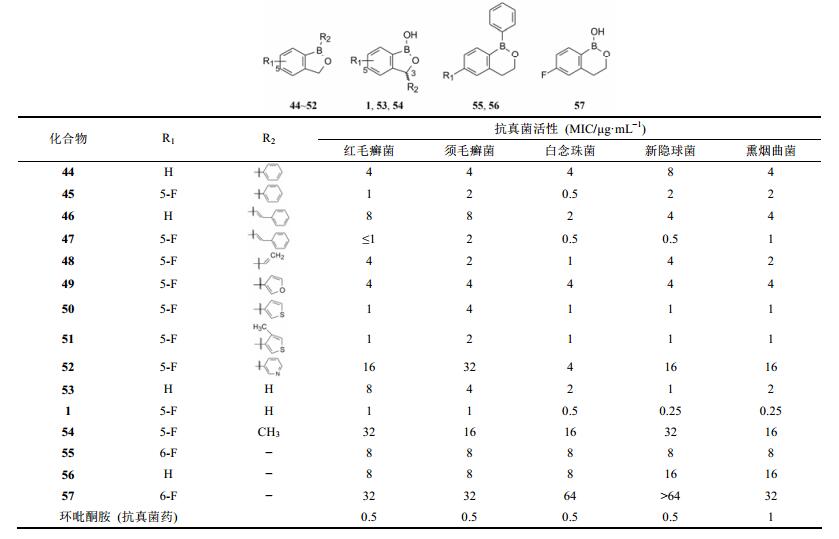
研究的目标添加了引起皮肤癣病(甲真菌病)的抗真菌作用。对新骨架化合物评价的模型为致病性真菌, 例如红毛癣菌、须毛癣菌、白念珠菌、新隐球菌和熏烟曲菌等, 活性强度用最低抑菌浓度(MIC)表示。
分析表 6的构效关系, 可简述如下: ① 以R2为苯环作为新骨架的起始物(44), 对真菌显示中等抑制活性。当在母核5位引入氟原子(45), 对所有真菌的抑制作用提高4~8倍。插烯物46及其氟代插烯物47分别与44和45相近, 提示氟原子具有增效作用, 因而后来设计的化合物都含有氟原子。② 苯环用乙烯基替换, 化合物48活性降低, 因而不是优化方向。③ 用呋喃(49)替换苯环, 活性减弱; 3-吡啶基化合物52尤其差。而噻吩(50、51)的活性与苯相当, 推测是亲脂性相似之故。④ 通式43的苯基被羟基置换, 没有F原子的53活性一般, 但5-F化合物(1)抗菌谱扩大, 活性也强于45。⑤ 3位引入甲基(54)活性显著降低, 推测该区域不宜被大体积占据或增加疏水性。⑥ 硼氧环扩为六员环的化合物活性都差, 提示硼氧五员环抗真菌具有特异性。
综合这一轮的结论是, 具有抗真菌作用化合物的特征是五员硼氧环为必要片段、3位不宜作取代、5位氟取代有利、1位连接硼原子的基团优选为苯基或羟基。
4.4 化合物45的1-苯基的优化鉴于化合物45为优选物之一, 对苯环A作不同取代, 合成的化合物列于表 7。
| 表 7 1-取代苯基化合物的抗真菌活性 |
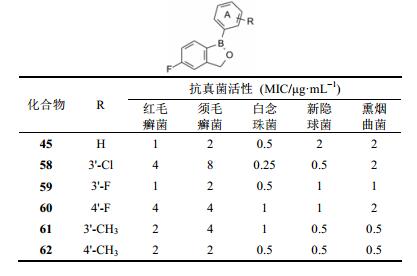
化合物58~62的苯环经不同位置的卤素或甲基取代, 抗菌谱和强度与45相近, 然而却出现了细胞毒作用, 因而终止了这一方向的优化。
4.5 化合物1的苯取代基的变换将化合物1的5-F变换成如图 4所列的不同取代基, 活性都下降; 氟原子由5位换到其他位置活性依然降低(数据省略)。

|
图 4 变换5-F合成的化合物均无改善 |
经多位点变换, 结果表明化合物1仍然是优胜的候选化合物(Baker SJ, Zhang YK, Akama T, et al. Discovery of a new boron-containing antifungal agent, 5-fluoro-1, 3-dihydro-1-hydroxy-2, 1-benzoxaborole (AN2690), for the potential treatment of onychomycosis. J Med Chem, 2006, 49: 4447−4450)。
5 候选物的确定和他伐硼罗上市化合物1的抗真菌谱和强度与已知抗真菌药环吡酮胺相近。进一步做机制研究, 证明是抑制亮氨酰转移RNA合成酶, 后者是真菌蛋白合成的关键酶, 体现了由表型评价到机制解析的研究过程。药代动力学研究表明, 趾(指)甲每日涂布5% (乙酸乙酯−丙二醇, 1:1) 溶液0.2 mL, 穿透趾甲作用为525 μg·cm−2。14天后的血浆最大浓度5.17 ng·mL−1, 血中半衰期28.5 h。经临床前研究表明有成药前景, 遂命名为他伐硼罗(tavaborole)进入开发阶段。临床研究表明, 局部涂抹他瓦硼罗的5%溶液治疗灰趾(指)甲是安全有效药物, 于2014年7月FDA批准在美国上市(Elewski BE, Aly R, Baldwin SL, et al. Efficacy and safety of tavaborole topical solution, 5%, a novel boron-based antifungal agent, for the treatment of toenail onychomycosis: results from 2 randomized phase-Ⅲ studies. J Am Acad Dermat, 2015, 73: 62−69)。
6 克立硼罗的研制综上, 研发者围绕有机硼酸酯的项目已获得以下结果:具有较强抗菌和抗炎活性的里程碑式化合物25, 以代号AN-0128拟进入临床研究; 化合物1以他伐硼罗名称上市成为新型抗甲癣药物; 积累了多样结构的含硼化合物库, 化合物库为新的抗过敏性皮炎药物提供了物质基础。
6.1 磷酸二酯酶4作为抗炎靶标文献报道磷酸二酯酶4 (PDE4) 的抑制剂可治疗过敏性皮炎(B umer W, Hoppmann J, Rundfeldt C, et al. Highly selective phosphodiesterase 4 inhibitors for the treatment of allergic skin diseases and psoriasis. Inflamm Allergy Drug Targets, 2007, 6: 17−26), 因而探索了含硼化合物同时对两个炎症靶标的抑制作用(抑制PDE4和促炎症细胞因子的产生)。
评价受试物抑制PDE4活性的方法是用人髓性白血病细胞制取半纯化的PDE4酶, 放入含3H-cAMP的cAMP底物, 加入不同浓度的受试物温孵, 终止反应后加入蛇毒核苷酸酶, 后者将生成的AMP水解为腺苷(含3H腺苷)。未水解的cAMP (连同3H-cAMP)固定在树脂上后, 上清液用液闪方法测定3H标记的腺苷含量, 计算化合物抑制PDE 4的IC50。与此同时还测定受试物对人外周血单核细胞释放细胞因子的抑制作用IC50 (如前述及)。
6.2 化合物对双靶标的活性和构效关系首先发现5-苯氧基化合物63具有抑制PDE4活性, IC50为6.8 μmol·L−1, 但不抑制(促)炎症因子的释放。因而以63为先导物进行改构, 即在末端苯环的不同位置引入取代基, 目标是对双靶标都呈现高活性的化合物。合成的化合物及其活性列于表 8。阳性对照药为PDE4抑制剂咯利普兰。
| 表 8 苯并硼氧戊烷化合物抑制PDE4和细胞因子的活性 |
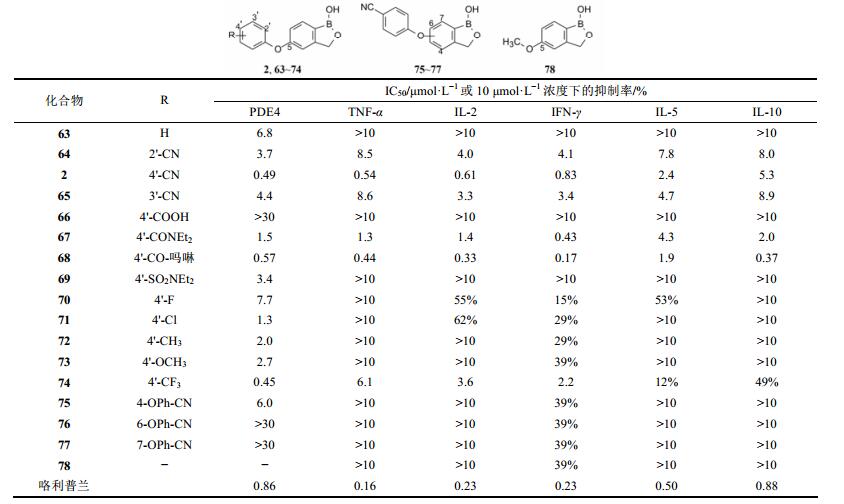
构效关系分析如下: ① 4'位为氰基(2)、吗啉甲酰基(68)和三氟甲基(74)的化合物对两类靶标都有抑制作用, 而且对PDE4的活性都强于咯利普兰。68抑制细胞因子的释放也强于咯利普兰, 但2不如咯利普兰。② 氰基的位置很重要, 移至2或3位(64、65)都使活性减弱。③ 4位被氯、甲基或甲氧基取代(71~73), 抑制PDE4的活性强于63, 但弱于2和68, 对细胞因子活性也不强。④ 4'-氰苯氧基连接在母核的5位很重要, 变换到4-、6-或7-位, 除化合物75对PDE4有弱作用外, 75~77对PED4和细胞因子都不呈现活性。末端苯环换成5-甲氧基(78)失去活性。
6.3 高活性化合物的动物实验对于两个优良化合物2和68用动物抗炎实验进行比较, 以佛波酯(phorbol ester)诱发小鼠耳肿胀模型, 局部涂抹受试物溶液, 评价消除肿胀和透入皮肤的能力。表 9列出了2和68的实验结果, 阳性对照药是地塞美松和咯利普兰。结果表明2与68有显著抑制动物耳部炎症的作用, 在同等剂量下2的活性略优于地塞美松, 强于3倍剂量的咯利普兰。实验还说明这两个硼化合物有良好的透皮作用。
| 表 9 化合物抑制佛波酯诱发小鼠耳肿胀的活性 |

综合2和68的药理活性和成药性前景, 化合物2抑制PDE 4和细胞因子的活性显著, 确定2为候选化合物, 定名克立硼罗(crisaborole), 经临床前和临床研究, 表明外用2%乳膏是治疗过敏性皮炎(湿疹)的有效药物, 美国FDA于2016年12月批准上市(Akama T, Baker SJ, Zhang YK, et al. Discovery and structure-activity study of a novel benzoxaborole anti-inflammatory agent (AN2728) for the potential topical treatment of psoriasis and atopic dermatitis. Bioorg Med Chem Lett, 2009, 19: 2129−2132)。
|
含硼元素的药物很少。早期作为消毒杀菌药的硼酸溶液, 现仅作为超市出售的清洗液。2003年上市治疗多发性骨髓瘤的硼替佐米是第一个有机硼药物, 为蛋白酶体抑制剂。本文介绍的姊妹药他伐硼罗和克立硼罗, 起始于大学的研究, 由Anacor公司研发成功。科学研究依靠兴趣与执着, 新药创制也是如此。从文献报道梳理的研发脉络显示, 起始于研究细菌的甲基转移酶抑制剂, 通过合成目标库发现有机硼酸酯的活性, 对硼化物同时评价抑制细胞因子的抗炎活性, 目标锁定为兼有细菌感染的过敏性皮炎, 研制外用有机硼化物避免了全身用药的潜在毒性。还研发了抑制真菌的硼化合物, 从而2014年治疗趾(指)甲癣的他伐硼罗成功上市。同时还研发针对磷酸二酯酶4和细胞因子的双靶标抗炎活性药物, 2016年上市了治疗湿疹的克立硼罗。坚持硼化合物为载体, 首创性地完成了两个治疗常见与多发的皮肤病药物。