 2017, Vol. 52
2017, Vol. 52



喹诺酮类药物 (quinolones, QNs) 是一类人工合成的具有1, 4-二氢-4-氧代喹啉-3-羧酸结构的化合物, 经过几十年的发展已成为临床上广泛使用的广谱、高效、低毒的一线抗菌药[1]。近年来研究发现, 喹诺酮类药物除了具有抗菌活性外, 还具有抗病毒[2]、抗贫血[3]、抗微生物[4]、抗肿瘤[5, 6]等多种药理活性。尤其是喹诺酮类药物的杀菌机制与现有依托泊苷、多柔比星等抗肿瘤药物杀死癌细胞的作用机制相似, 都是通过抑制拓扑异构酶Ⅱ介导的DNA解旋反应来干扰DNA复制并导致细胞死亡[7], 这使得喹诺酮类药物有望成为一类新型的抗肿瘤药物。事实上, 在转化喹诺酮类抗菌剂到抗肿瘤药物上已经做了大量的探索性研究[8-11], 目前已设计合成了数百个具有抗肿瘤活性的喹诺酮类衍生物, 但遗憾的是这些化合物由于自身抗肿瘤作用弱、毒性高、生物利用度差等缺点限制了其进一步开发。因此, 寻找新的修饰方法对喹诺酮类药物进行改造, 以期得到抗肿瘤活性更强、毒性更低的衍生物具有重要的研究意义。
研究表明, 肿瘤的发生、发展与基因水平病变有着密切关系。组蛋白乙酰化转移酶 (histone acetyl transferases, HATs) 和组蛋白去乙酰化酶 (histone deacetylases, HDACs) 是调控基因转录与表达的两个关键酶家族[12]。在癌细胞中HDACs过量表达使得原有基因在转录、表达中的组蛋白过度去乙酰化, 增强了组蛋白与DNA的结合能力, 促使核小体结构变紧, 使得一些调控细胞周期和影响细胞增殖的分子无法与DNA结合, 阻止基因转录, 最终导致肿瘤的发生[13], 尤其是HDAC1、HDAC2和HDAC6这3种亚型在多种肿瘤细胞中均过表达[14-16]。组蛋白去乙酰化酶抑制剂 (histone deacetylase inhibitor, HDACi) 能够抑制肿瘤细胞内HDACs活性, 增加肿瘤细胞内组蛋白N-末端赖氨酸残基的乙酰化程度, 重新活化受抑制的抑癌基因, 诱导肿瘤细胞生长阻滞、分化、凋亡[17]。因此, HDACs是近年来抗肿瘤研究的一个新兴靶点, HDACi也成为国内外抗肿瘤药物研究热点。自FDA批准的第一个HDACs抑制剂伏立诺他 (SAHA) 用于治疗外周T细胞淋巴瘤后[18], 已报道了数百种HDACi, 例如将SAHA的苯环用1, 3, 4-噻二唑修饰后, 所得化合物 (1A, 图 1) 仍保留了较强的HDACs抑制活性[19, 20]。目前报道的HDACi的结构大致可以分为四部分 (图 1):表面识别区 (CAP)、连接基团 (CU)、疏水连接区 (HS) 和锌离子结合区 (ZBG)。当HDACi与HDACs相互作用后, 表面识别区位于酶催化口袋的入口处, 由于该区域的空间面积比较大, 而HDACi表面识别区仅占据了小部分面积[21], 因此将HDACi表面识别区拼接另一抗肿瘤药物不仅有利于增强HDACi与酶的结合, 提高其抗癌活性, 而且构建的新化合物还可能具有双靶向性。因此, 本文拟将HDACi拼接到喹诺酮类药物左氧氟沙星 (化合物1B, 图 1) 上来构建左氧氟沙星-HDACi缀合物, 以期得到新型、高效、低毒的左氧氟沙星衍生物。

|
Figure 1 The structures of SAHA, thiadiazole HDACi and levofloxacin |

|
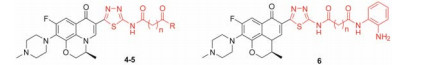
Scheme1 Synthetic route of levofloxacin-HDACi conjugates 4-6. Reagents and conditions: (a) Aminothiourea, polyphosphoric acid, 120 ℃, 24 h; (b) THF, rt, 2 h; (c) ClCO2Et, Et3N, THF, 0 ℃, 15 min and then NH2OH·HCl, KOH, MeOH, rt, 1 h; (d) (ⅰ) (C2H5)3N, BOP reagent, dry DMF, 0.5 h, rt; (ⅱ) o-Phenylendiamine, dry DMF, rt, 12 h |
已有喹诺酮类药物的结构改造发现, 喹诺酮类药物C-3位羧基用杂环取代能够提高其抗肿瘤活性[22, 23]。因此, 本研究拟将噻二唑类HDACi引入到喹诺酮类药物左氧氟沙星的C-3位构建含有噻二唑杂环连接的左氧氟沙星-HDACi缀合物 (合成路线1, 化合物5); 同时, 为了进一步研究缀合物的构效关系, 也合成了羧酸类 (缀合物4) 和苯甲酰胺类缀合物 (化合物6)。测试了所合成的目标缀合物对HDACs的抑制活性和体外抗肿瘤活性。结果表明, 目标缀合物均呈现了较强的HDACs抑制活性和体外抗肿瘤活性, 其中肟酸类缀合物对HDACs的抑制活性强于羧酸和苯甲酰胺类衍生物, 尤其是肟酸缀合物5d对HDAC1和HDAC6的抑制活性强于阳性药物SAHA, 并对SW620、MGC-803、PC-3、NCIH460、MCF-7和HepG2 6种肿瘤细胞抑制活性最强。
目标缀合物4~6的合成过程见合成路线1。以左氧氟沙星为原料, 用多聚磷酸为脱水剂经一锅法合成2-氨基-1, 3, 4-噻二唑左氧氟沙星2, 接着与相应的酸酐3反应制得羧酸系列左氧氟沙星-HDACi缀合物4, 再用羟胺将缀合物4的羧基转化成异羟肟酸得到了相应的目标缀合物5; 另一方面, 将羧酸缀合物4直接与邻苯二胺反应制得苯甲酰胺类目标缀合物6。所合成的18个目标缀合物均未见文献报道。
结果与讨论 1 合成部分目标缀合物的结构经1H NMR、13C NMR和HR-MS分析确证。其收率、理化常数及波谱数据见表 1、2。
| Table 1 Physical constants and HR-MS of synthesized compounds 4-6 |
| Table 2 The NMR data of target compounds 4-6 |
目标缀合物对HDACs的抑制作用见表 3, 可以发现, 所合成的18个左氧氟沙星-HDACi缀合物对HDAC1、HDAC2和HDAC6均有较强的抑制活性, 这些结果说明目标缀合物对组蛋白去乙酰化酶有较强的靶向性。从目标缀合物的结构上看, 缀合物是将左氧氟沙星取代了SAHA的苯环, 而在HDACs的酶活性口袋中SAHA的苯环仅占据了活性口袋入口处的较小空间, 用左氧氟沙星取代苯环后增加了化合物与酶的相互作用。因此, 缀合物显示出了较强的HDACs抑制活性。但是, 缀合物HDACi单元的疏水连接链长度对HDACs的抑制活性有显著的影响。其中连接链为6个亚甲基 (n = 6) 对HDACs抑制活性最佳, 缩短 (n = 3、4、5) 或延长 (n = 7、8) 链长均减弱对HDACs的抑制活性。例如在羧酸系列缀合物4中, 缀合物4d (n = 6) 对HDACs的抑制活性强于缀合物4a (n = 3)、4b (n = 4)、4c(n = 5)、4e (n = 7) 和4f (n = 8), 同时这个趋势在肟酸系列和苯甲酰胺系列也存在。此外, 目标缀合物中锌离子结合基团 (羧酸、肟酸、苯甲酰胺) 对HDACs的抑制活性也有较大的影响, 其中肟酸系列缀合物5对HDAC1、HDAC2和HDAC6的抑制活性明显地强于相应的羧酸系列缀合物4和苯甲酰胺系列缀合物6。尤其是肟酸缀合物5d在所有化合物中对HDAC1、HDAC2和HDAC6呈现出了最强的抑制活性, 其IC50分别为0.031、0.041、0.019 μmol·L-1, 其对HDAC1和HDAC6的抑制活性略强于阳性药物SAHA。
| Table 3 In vitro inhibition of HDAC1, HDAC2 and HDAC6. n = 3, x± s |
在HDACs抑制实验中, 目标缀合物选择对HDAC1和HDAC6抑制活性最强的缀合物5d进行分子对接, 并将最佳对接构象分子用Pymol进行绘图, 其对接结果如图 2所示。在HDAC1和HDAC6的口袋中, 缀合物5d表面识别区左氧氟沙星单元均位于活性口袋的入口处 (图 2A, B), 较SAHA表面识别区苯环在活性口袋的入口处占据了更多的空间, 因此引入左氧氟沙星增强了SAHA与HDAC1和HDAC6的相互作用。同时, 缀合物5d的脂肪链与酶活性口袋的疏水通道壁有较强的疏水作用 (图 2A, B), 并且肟酸基团能够与酶活性口袋底部的氨基酸残基以及Zn2+相互作用。但是, 肟酸基团与HDAC1和HDAC6活性口袋底部的氨基酸残基和锌离子的作用方式却不相同。在HDAC1中, 肟酸基团是与H178和Y303两个氨基酸残基相互作用形成了3个氢键, 并且肟酸基团的羰基 (3.3Å) 和羟基 (1.9Å) 能够与氨基酸残D176 (2.0Å, 2.9Å)、H178 (2.3Å)、D264 (3.0Å) 一起与Zn2+形成六配位 (图 2C); 而在HDAC6中, 缀合物5d的肟酸基团与氨基酸残基Y782相互作用形成了两个氢键, 同时肟酸基团的羰基氧 (3.2Å) 和羟基氧 (2.5Å) 能够与氨基酸残基D649 (2.1Å)、H651 (1.9Å)、D742 (2.7Å) 一起与Zn2+形成五配位 (图 2D)。此外, 缀合物5d左氧氟沙星骨架的羰基和连接基团噻二唑的氮原子在HDAC6中能够分别与氨基酸残基Y782和F679形成氢键 (图 2D)。

|
Figure 2 The predicted binding modes of compound 5d-HDAC1 and HDAC6. (A) Molecular surface of the HDAC1 binding pocket with docked compound 5d. (B) Molecular surface of the HDAC6 binding pocket with docked compound 5d. (C) Docking poses of HDAC1-5d, which can form hydrogen bonds with residues H178, Y303, and which can coordinate the zinc ion with residues D176, H178, D264. (D) Docking poses of HDAC6-5d, which can form hydrogen bonds with residuesF679, Y782, and which can coordinate the zinc ion with residues D649, H651, D742. Distances are given in Å |
体外抗肿瘤测试结果 (表 4) 表明, 18个目标左氧氟沙星-HDACi缀合物对SW620、MGC-803、PC-3、NCIH460、MCF-7和HepG2这6种肿瘤细胞有较强的抑制作用, 其IC50值均小于17.0 μmol·L-1。由于在左氧氟沙星引入了具有抗肿瘤活性的HDAC抑制剂SAHA, 使得左氧氟沙星缀合物不仅能够通过抑制拓扑异构酶Ⅱ来发挥抗肿瘤活性, 还能够通过抑制HDACs来发挥抗肿瘤活性, 具有双重抗肿瘤作用, 因此合成的缀合物抗肿瘤活性显著地强于先导化合物左氧氟沙星 (IC50 > 67 μmol·L-1)。同时, 这些缀合物对胃癌MGC-803细胞呈现出了更强的抑制活性, 如缀合物5b~5e、6c~6e对MGC-803的抑制活性强于阳性药物SAHA, 其中缀合物5c、5d和6d抑制MGC-803的IC50达到了10-7 mol·L-1水平。在HDACs抑制活性中, 肟酸系列缀合物 (化合物5) 的HDACs抑制活性强于羧酸类 (化合物4) 和苯甲酰胺类缀合物 (化合物6)。同样的, 在抗肿瘤活性中, 肟酸的缀合物5抗癌活性也强于相应的羧酸类缀合物4和苯甲酰胺类缀合物6。特别是对HDACs有最强抑制活性的肟酸缀合物5d对6种肿瘤细胞也展现出了最强的抑制活性, 其对MGC-803、NCIH460、MCF-7和HepG2抑制活性是阳性药物SAHA的3.1倍以上。但是, 与先导化合物左氧氟沙星相比, 引入SAHA后能明显地提高左氧氟沙星的抗肿瘤活性, 而与SAHA相比, 这些缀合物并没有显著地提高抗肿瘤活性。在拓扑异构酶Ⅱ抑制活性中 (图 3), 发现这些缀合物对TopⅡ仅展现出了中等的抑制活性, 因此目标缀合物通过抑制TopⅡ途径来提高SAHA的抗肿瘤活性效果并不显著。这些结果也说明了目标缀合物的抗肿瘤活性主要源于抑制HDACs。

|
Figure 3 Human DNA Topo Ⅱ inhibitory activity of conjugates 5a-5f at the concertration of 50 μmol·L-1. Lane D: pBR322 DNA only; Lane T: pBR322 DNA + Topo Ⅱ; Lane E: pBR322 DNA + Topo Ⅱ + Etoposide; Lane 4-9: pBR322 DNA + Topo Ⅱ + each of compounds 5a-5f |
| Table 4 Whole cell antiproliferative activity of target compounds. n = 3, x± s |
从表 4抗肿瘤活性中发现, 目标缀合物对胃癌MGC-803细胞有较强的抑制活性, 为了进一步评价目标缀合物的毒性, 用正常的胃黏膜上皮细胞GES-1作为测试细胞株来检测缀合物的毒性。从表 4中可以发现, 总的来说这类缀合物基本没有毒性 (IC50 > 81 μmol·L-1), 而阳性药物SAHA (IC50 = 5.8 ± 0.48 μmol·L-1) 却展现出了一定的毒性。
5 拓扑异构酶Ⅱ抑制活性对体外HDACs抑制活性和抗肿瘤活性较好的5a~5f, 采用超螺旋pBR322质粒DNA解螺旋电泳实验测试了目标化合物在50 μmol·L-1浓度下对拓扑异构酶Ⅱ (TopⅡ) 的抑制活性, 以依托泊苷 (etoposide) 为阳性对照药。结果显示 (图 3), 化合物5a~5f表现出了中等的TopⅡ抑制活性, 其中化合物5d对TopⅡ的抑制活性强于5a~5c和5e~5f, 但是低于阳性药物依托泊苷。
6 小结通过将具有优秀抗肿瘤活性的噻二唑组蛋白去乙酰化酶抑制剂引入到左氧氟沙星中, 合成了18个左氧氟沙星-HDACi缀合物。经过测试这些目标缀合物对HDACs的抑制活性发现, 其对HDAC1、HDAC2和HDAC6均有较强的抑制活性, 其中链长为6个亚甲基 (n = 6)、锌离子结合基团为肟酸的缀合物5d展现出了最强的HDACs抑制活性, 其对HDAC1和HDAC6的抑制活性略强于阳性药物SAHA。分子对接研究表明, 左氧氟沙星与HDACi的连接基团噻二唑对缀合物HDAC6选择性起着重要作用。体外抗瘤活性实验发现, 目标缀合物对SW620、MGC-803、PC-3、NCIH460、MCF-7和HepG2这6种肿瘤细胞均有较强的抑制活性, 特别是对胃癌MGC-803细胞有更强的抑制活性和选择性, 其中缀合物5d对MGC-803的抑制活性最强, 分别是先导化合物左氧氟沙星和SAHA的90和4.8倍; 此外, 缀合物5d对正常的胃黏膜细胞GES-1没有毒性, 对拓扑异构酶Ⅱ也有抑制作用。这些结果说明将HDACi拼接到喹诺酮类药物上能够提高其抗肿瘤活性, 这为今后发展新型、高效、低毒的喹诺酮类抗肿瘤药物提供了新的思路。
实验部分BrukerAM-400 Hz型核磁共振仪 (德国Bruker公司); X-5显微熔点测定仪 (上海越众设备有限公司); FA1104电子天平 (上海上平仪器有限公司); B11-3温度数显恒温磁力搅拌器 (上海司乐仪器有限公司); Q Exactive高分辨质谱仪 (美国Coring公司); 37 ℃ CO2培养箱 (美国Thermo公司); CKX31型倒置显微镜 (奥林巴斯公司); ELx800通用酶标仪 (美国BioTek公司)。
左氧氟沙星、伏立诺他 (vorinostat, SAHA)、多聚磷酸、氨基硫脲、BOP试剂、邻苯二胺、盐酸羟胺等试剂均购自百灵威科技有限公司 (质量分数≥ 95%); 实验所用的其他试剂购自国药集团化学试剂有限公司 (质量分数≥ 95%); DMEM、MEM培养基、胎牛血清购自Hyclone公司; SW620、MGC-803、PC-3、NCIH460、MCF-7和HepG2细胞株购自武汉大学细胞典藏中心。
1 化学合成 1.1 2-氨基-1, 3, 4-噻二唑左氧氟沙星 (2) 的合成将左氧氟沙星1 (4.155 g, 11.5 mmol)、氨基硫脲 (1.048 g, 11.5 mmol) 和多聚磷酸 (20 mL) 混合后将反应液加热至120 ℃, 反应24 h, 将反应液冷却到室温后倒入100 mL的冰浴中, 用乙酸乙酯 (3 × 30 mL) 萃取, 有机层用无水Na2SO4干燥, 减压脱除溶剂得到粗产物, 经柱色谱纯化 (石油醚-乙酸乙酯=1:1) 得到了2-氨基-1, 3, 4-噻二唑左氧氟沙星2。黄色固体, 产率: 77%, mp 175~177 ℃; 1H NMR (400 MHz, Acetone-d6) δ: 8.86 (s, 1H), 7.86 (d, J = 13.2 Hz, 1H), 4.66~4.52 (m, 3H), 3.37~3.24 (m, 4H), 2.64~2.55 (m, 4H), 2.34 (s, 3H), 1.65 (d, J = 7.2 Hz, 3H)。
1.2 羧酸类左氧氟沙星-HDACi缀合物 (4) 的合成氩气保护下, 将戊 (己、庚、辛、壬、癸) 二酸酐3 (2.4 mmol) 溶于无水THF中 (30 mL), 加入2-氨基-1, 3, 4-噻二唑左氧氟沙星2 (1.083 g, 2.6 mmol) 后, 在室温下继续反应2 h, 减压脱除溶剂得到粗产物, 经柱色谱纯化 (二氯甲烷-甲醇=10:1) 得到相应的羧酸类缀合物4。
1.3 肟酸类左氧氟沙星-HDACi缀合物 (5) 的合成氩气保护下, 将羧酸化合物4 (1.2 mmol)、氯甲酸乙酯 (206 mg, 1.9 mmol)、三乙胺 (131 mg, 1.3 mmol) 溶解到10 mL THF中, 0 ℃下搅拌15 min后向反应液中加入10 mL新制备的羟胺溶液, 将反应液缓慢升至室温后继续反应2 h, 减压除去溶剂得到粗产物, 经柱色谱纯化 (二氯甲烷-甲醇=25:1~9:1) 得到了异肟酸化合物5。羟胺溶液的制备:将盐酸羟胺 (333 mg, 4.8 mmol) 和氢氧化钾 (269 mg, 4.8 mmol) 加入到甲醇 (10 mL) 中, 升温至40 ℃反应15 min后, 析出大量的沉淀, 将反应液冷却至室温, 滤除沉淀, 所得滤液即为新制备的羟胺溶液。
1.4 苯甲酰胺类左氧氟沙星-HDACi缀合物 (6) 的合成氩气保护下, 将羧酸缀合物4 (0.6 mmol)、苯并三氮唑-1-基氧基三 (二甲基氨基) 磷鎓六氟磷酸盐 (BOP试剂, 575 mg, 1.3 mmol) 和三乙胺 (394 mg, 3.9 mmol) 溶解到无水DMF (15 mL) 中, 室温反应30 min后, 加入邻苯二胺 (76 mg, 0.7 mmol), 继续反应12 h, 向反应液中加入20 mL蒸馏水, 用乙酸乙酯 (3 × 35 mL) 萃取, 有机层用无水Na2SO4干燥, 脱除溶剂, 经柱色谱纯化 (二氯甲烷-甲醇=25:1~9:1) 得到苯甲酰胺类缀合物6。
2 HDACs抑制活性实验使用HDAC试剂盒测试左氧氟沙星-HDACi缀合物对HDAC1、HDAC2、HDAC6的抑制活性, 以SAHA为阳性对照。按照HDAC试剂盒操作说明书在测试板中每孔加入BSA、HDAC荧光底物、HDAC酶 (HDAC1、HDAC2、HDAC6) 和不同浓度的待测物, 其浓度梯度分别为: 1×10-10、1×10-9、1×10-8、1×10-7、1×10-6、1×10-5 mol·L-1。将测试板在37 ℃下反应30 min后, 每孔再加入HDAC Developer并继续在37 ℃下放置15 min。使用酶标仪在359和440 nm波长测定每孔的荧光值, 分析实验结果, 并计算出IC50, 以此判断合成的分子探针对组蛋白去乙酰酶的靶向性。
3 分子对接HDAC1晶体结构 (PDB code: 4BKX) 由Protein Data Bank下载, HDAC6的晶体结构目前还未解析出来, 按照参考文献以HDAC8晶体结构 (PDB code: 1T69) 为模板对HDAC6进行同源建模[24-26]。通过ChemBio 3D ultra 14.0和Autodock4.2D中的Ligand模块进行小分子结构的预处理。对接软件使用Autodock4.2D, 利用晶体结构中的配体定义口袋盒子, 对接盒子边长设置为30 Å, 使用半经验自由能进行评价, 拉马克遗传算法循环100次, 其余参数保持默认。
4 抗增殖活性实验选取结肠癌SW620、胃癌MGC-803、前列腺癌PC-3、大细胞肺癌NCIH460、乳腺癌MCF-7和肝癌HepG2细胞为测试细胞株, 正常的胃黏膜上皮细胞GES-1为对照细胞。以SAHA为阳性对照药, 采用CCK-8试剂盒对左氧氟沙星-HDACi缀合物进行抗肿瘤活性评价。取对数生长期的测试细胞株悬浮于含10%胎牛血清的培养基中, 铺至96孔培养板中。待细胞完全贴壁后, 弃去原培养液, 加入10 μL含有测试药物的培养液培养48 h, 弃去原培养液, 每孔加入10 μL CCK-8溶液, 在培养箱中继续孵育4 h, 用酶标仪在490 nm波长测定每孔的吸光度 (OD) 值, 分析实验结果, 并计算出IC50。
5 拓扑异构酶Ⅱ抑制活性实验按TopⅡ试剂盒的说明书, 将5 × complete buffer液 (5 μL)、TopⅡ (8 U)、pBR322质粒 (0.25 μg) 和测试化合物 (0.2 μL, 50 μmol·L-1) 用蒸馏水定容到20 μL, 在37 ℃下放置20 min后, 在反应液中继续加入5% SDS (2 μL) 和蛋白酶K溶液 (2 μL, 1 mg·mL-1)。将反应液在37 ℃下继续反应30 min, 加入10 × loading buffer液 (2 μL) 后在70 ℃水浴中反应2 min, 加入CIA (20 μL, 苯酚-氯仿-异戊醇= 25:24:1), 振荡后离心, 取上清液上样, 90 V电压进行电泳2 h。取出凝胶后置于UV成像系统中成像。
| [1] | Jiang XL, Cui YB, Cao SH. Progress in research on qui-nolone antibacterials[J]. Chin J Antibiot (中国抗生素杂志), 2011, 36: 255–263. |
| [2] | Khan IA, Siddiqui S, Rehmani S, et al. Fluoroquinolones inhibit HCV by targeting its helicase[J]. Antivir Ther, 2012, 17: 467–476. |
| [3] | Murray JK, Balan C, Allgeier AM, et al. Dipeptidyl-quinolone derivatives inhibit hypoxia inducible factor-1α prolyl hydroxylases-1, -2, and-3 with altered selectivity[J]. J Comb Chem, 2010, 12: 676–686. DOI:10.1021/cc100073a |
| [4] | Patel D, Kumari P, Patel N. Synthesis and biological evaluation of some thiazolidinones as antimicrobial agents[J]. Eur J Med Chem, 2012, 48: 354–362. DOI:10.1016/j.ejmech.2011.11.041 |
| [5] | Li T, Gao LZ, Xie YS, et al. Synthesis and anti-proliferative activity of fluoroquinolone C-3 fused heterocyclic α, β-unsaturated ketones derived from ciprofloxacin[J]. Acta Pharm Sin (药学学报), 2015, 50: 569–573. |
| [6] | Gao LZ, Xie YS, Yan Q, et al. Synthesis and anti-proliferative activity of fluoroquinolone (rhodamine unsaturated ketone) amide derivatives[J]. Acta Pharm Sin (药学学报), 2015, 50: 1008–1012. |
| [7] | Katsarou ME, Efthimiadou EK, Psomas G, et al. Novel copper (Ⅱ) complex of N-propyl-norfloxacin and 1, 10-phe-nanthroline with enhanced antileukemic and DNA nuclease activities[J]. J Med Chem, 2008, 51: 470–478. DOI:10.1021/jm7013259 |
| [8] | Hu GQ, Hou LL, Wang GQ, et al. Design, synthesis and antitumor activity of fluoroquinolone C-3 heterocycles:bis-oxadiazole methylsulfide derivatives derived from ciprofloxacin[J]. Acta Pharm Sin (药学学报), 2012, 47: 1017–1022. |
| [9] | Wang FH, Wang K, Shen JH, et al. Design, synthesis and antiproliferative activity of 3-hydroxy-4-quinolone[J]. J China Pharm Univ (中国药科大学学报), 2014, 45: 286–292. |
| [10] | Shang HJ, Yan Q, Wu SM, et al. Synthesis and antitumor activity of novel oxadizole derivatives of enrofloxacin[J]. J Shenyang Pharm Univ (沈阳药科大学学报), 2015, 32: 604–608. |
| [11] | Gao Z, Tao L, Xie YS, et al. Design, synthesis, antibac-terial and anti-cell proliferation activities of[J]. Acta Pharm Sin (药学学报), 2015, 50: 332–336. |
| [12] | Smith BC, Denu JM. Chemical mechanisms of histone lysine and arginine modifications[J]. Biochim Biophys Acta, 2009, 1789: 45–57. DOI:10.1016/j.bbagrm.2008.06.005 |
| [13] | Lai MJ, Huang HL, Pan SL, et al. Synthesis and biological evaluation of 1-arylsulfonyl-5-(N-hydroxyacrylamide) indoles as potent histone deacetylase inhibitors with antitumor activity in vivo[J]. J Med Chem, 2012, 55: 3777–3791. DOI:10.1021/jm300197a |
| [14] | Wagner FF, Olson DE, Gale JP, et al. Potent and selective inhibition of histone deacetylase 6(HDAC6) does not require a surface-binding motif[J]. J Med Chem, 2013, 56: 1772–1776. DOI:10.1021/jm301355j |
| [15] | Lee HY, Tsai AC, Chen MC, et al. Azaindolylsulfona-mides, with a more selective inhibitory effect on histone deacetylase 6 activity, exhibit antitumor activity in colorectal cancer HCT116 cells[J]. J Med Chem, 2014, 57: 4009–4022. DOI:10.1021/jm401899x |
| [16] | Tang C, Li CH, Zhang SL, et al. Novel bioactive hybrid compound dual targeting estrogen receptor and histone deacetylase for the treatment of breast cancer[J]. J Med Chem, 2015, 58: 4550–4572. DOI:10.1021/acs.jmedchem.5b00099 |
| [17] | Yao YW, Yao HQ, Jiang S, et al. Progress in clinical study of histone deacetylases inhibitors as anticancer agents[J]. Chin J New Drugs (中国新药杂志), 2013, 22: 294–299. |
| [18] | Marks PA. Discovery and development of SAHA as an anticancer agent[J]. Oncogene, 2007, 26: 1351–1356. DOI:10.1038/sj.onc.1210204 |
| [19] | Guan P, Sun F, Hou X, et al. Design, synthesis and preliminary bioactivity studies of 1, 3, 4-thiadiazole hydroxamic acid derivatives as novel histone deacetylase inhibitors[J]. Bioorg Med Chem, 2012, 20: 3865–3872. DOI:10.1016/j.bmc.2012.04.032 |
| [20] | Guan P, Hou X, Wang L, et al. Improved antiproliferative activity of 1, 3, 4-thiadiazole-containing histone deacetylase (HDAC) inhibitors by introduction of the heteroaromatic surface recognition motif[J]. Bioorg Med Chem, 2014, 22: 5766–5775. DOI:10.1016/j.bmc.2014.09.039 |
| [21] | Tan YM, Huang WY, Yu NF. Structure-activity relationships of histone deacetylase inhibitors[J]. Acta Pharm Sin (药学学报), 2009, 44: 1072–1083. |
| [22] | Xie SQ, Chen YS, Wang GQ, et al. Part Ⅳ. Synthesis and antitumor evaluation of s-triazolothiadiazines and pyrazolo s-triazoles derived from criproxacin[J]. Acta Pharm Sin (药学学报), 2012, 47: 66–71. |
| [23] | Gao LZ, Xie YS, Li T, et al. Synthesis, antitumor activity and SAR of C-3 oxadiazole sulfanylacetylhydrazone-substituted fluoroquinolone analogues[J]. Acta Pharm Sin (药学学报), 2014, 49: 1694–1698. |
| [24] | Itoh Y, Suzuki T, Miyata N. Isoform-selective histonedeacetylase inhibitors[J]. Curr Pharm Des, 2008, 14: 529–544. DOI:10.2174/138161208783885335 |
| [25] | No author. Activities at the universal protein resource (UniProt)[J]. Nucleic Acids Res, 2014, 42: D191–D198. DOI:10.1093/nar/gkt1140 |
| [26] | Senger J, Melesina J, Marek M, et al. Synthesis and biological investigation of oxazole hydroxamatesas highly selective histone deacetylase 6(HDAC6) inhibitors[J]. J Med Chem, 2016, 59: 1545–1555. DOI:10.1021/acs.jmedchem.5b01493 |