 2017, Vol. 52
2017, Vol. 52



泛素-蛋白酶体系统 (ubiquitin-proteasome system, UPS) 是真核细胞中与蛋白质质量控制和细胞稳态维持相关的一个关键ATP-依赖性降解系统, 可以特异性识别和降解多种蛋白质底物, 特别是不稳定或变性、错误折叠的蛋白质。该系统中, 目标蛋白质依次经过泛素活化酶E1、泛素结合酶E2和泛素连接酶E3作用, 共价修饰上多聚泛素链从而被20S核心颗粒 (core particle, CP) 与19S调节颗粒 (regulatory particle, RP) 结合形成的26S蛋白酶体识别并降解为短肽[1-3]。其中, CP是由两个外层α环和两个内层β环堆叠而成的720 kDa中空圆桶状亚复合体, 每一层环由7个密切相关亚基组成并可表示为α1-7β1-7β1-7α1-7。蛋白酶体活性位点即位于CP内腔, 由β亚基Thr1形成独特的单个残基催化位点, 7种β亚基中有3种由于Thr1的存在具有催化活性: β1表现胱冬肽酶样 (caspase-like, C-L) 活性, β2表现胰蛋白酶样 (trypsin-like, T-L) 活性, β5表现胰凝乳蛋白酶样 (chymotrypsin-like, ChT-L) 活性并主要负责底物降解[4, 5]。由α环7个α亚基N-端参与形成的闭合门样结构会限制底物进入CP内腔到达活性位点, 结合至CP的RP则通过其亚基C-端与α环互作诱导开门构象形成[6, 7]。700 kDa的蛋白酶体激活因子 (proteasome activator MW 700, PA700) 19S RP是真核蛋白酶体最保守的调节颗粒, 结合至CP一端或两端分别形成26S和30S蛋白酶体[1, 7]。26S蛋白酶体中的19S亚复合体可分为底和盖两部分。底的一个关键组件是Rpt (regulatory particle AAA ATPase) 1-6亚基形成的异源六聚体Rpt环, Rpt环关联的Rpn (regulatory particle non-ATPase) 1、2则识别蛋白酶体常驻泛素受体Rpn10、13;盖结构中Rpn3、5-7、9和12形成一个马蹄铁样结构并围绕由蛋白酶体必需去泛素化酶Rpn11与另一个亚基Rpn8形成的异二聚体。目前认为, Rpn10和Rpn13联合识别底物的泛素链标签, 一旦泛素化底物与Rpt环结合并发生ATP依赖的解折叠, Rpn11将移除泛素链重循环泛素同时促进底物易位CP内腔, 且该过程受到不同层次变构调节的控制[8, 9]。高等真核生物中存在组织特异性蛋白酶体, CP通过从头组装过程中亚基替代的方式形成变种。如脊椎动物免疫细胞等多种细胞受IFN-γ (interferon-gamma) 诱导, CP β1、β2和β5分别被裂解特异性改变了的β1i、β2i和β5i替代, 对应于组成型蛋白酶体核心颗粒 (constitutive proteasome core particle, cCP) 形成免疫蛋白酶体核心颗粒 (immunoproteasomes core particle, iCP)。免疫蛋白酶体能够更加有效地促进MHC-Ⅰ抗原提呈从而对细胞毒性T细胞反应触发十分重要[1, 10, 11]。此外, 脊椎动物能够编码一种胸腺特异的CP β亚基β5t来替代iCP中β5i从而形成胸腺蛋白酶体核心颗粒 (thymo proteasomes core particle, tCP)。胸腺蛋白酶体能够促进CD8+ T细胞阳性选择和功能发育[2]。
UPS途径通过对细胞内发挥调节作用的蛋白质的特异性靶向破坏, 在许多重要的细胞活动中扮演关键角色, 已在数种疾病中发现其功能异常。如细胞周期蛋白及周期蛋白依赖性激酶抑制因子没有UPS介导的及时降解将引起细胞周期阻滞和细胞凋亡[12]。也正是这一发现推测蛋白酶体抑制剂 (proteasome inhibitors, PIs) 可以阻止肿瘤细胞增殖。考虑到蛋白酶体在确保正常细胞功能的几乎所有细胞过程而不仅仅是细胞周期进程中都扮演重要角色, 其在某一时间点的抑制被认为是非特异的, 不值得作为药物靶标进一步研究。然而, 肿瘤细胞比正常细胞对PIs远为敏感的发现为发展其靶向药物铺平了道路[13]。基因组不稳定性和快速增殖性使得肿瘤细胞更加依赖于蛋白酶体移除错误折叠或损伤的蛋白质, 事实上, 原发性肿瘤中蛋白酶体水平和活性远高出正常组织[14]。PIs选择性诱导肿瘤细胞凋亡的同时有助于提高对放、化疗的敏感性, 26S蛋白酶体已成为本世纪最有前途的抗癌药物靶点之一。目前, FDA (US Food and Drug Administration) 批准临床/临床前研究的PIs多集中在对新诊断和复发/难治性多发性骨髓瘤 (multiple myeloma, MM) 的治疗, 这至少部分与MM细胞由于高IgG生物合成率而对蛋白酶体的高依赖性有关[15, 16]。除了作为抗癌试剂, PIs在炎症和自身免疫性疾病、器官移植及再灌注损伤等疾病的动物模型中也表现出积极作用[16, 17]。
2 20S CP靶向的PIs26S蛋白酶体活性位点位于CP内腔。CP作为有价值的药物作用靶点, 其靶向化合物已得到广泛和深入的研究[15-18]。传统CP靶向抑制剂并未表现出对cCP/iCP具有选择性作用, 至少部分解释了这类药物具有的某些不良反应。随着CP及其变种晶体结构的明确, 利用它们活性位点间结构差异开发出的具有cCP/iCP选择靶向性的抑制剂得以发展[19]。此外, 选择性靶向病原体如恶性疟原虫 (Plasmodium falciparum, P. falciparum) 而非宿主细胞CP的抑制剂对依赖于蛋白酶体功能存活和繁殖的致病微生物也有望成为有潜力的治疗策略[20]。
2.1 传统20S CP靶向抑制剂 (对cCP/iCP无选择性靶向的PIs) 2.1.1 共价作用型CP靶向的多数抑制剂都是通过模拟酶的一个底物在Thr1相邻位置形成β折叠样接触从而靶向活性位点, 其中许多还同时依赖于分子另一端一个亲电反应活性基团与Thr1共价互作[21, 22]。为得到具有高效力、高选择性和良好药理特性的该类抑制剂, 靶向分子中亲电头部被大量开发。早期发展的CP靶向分子有醛肽类 (peptide aldehydes)[23, 24]、硼酸肽类 (peptide boronates)[13, 18]、环氧酮肽类 (peptide epoxyketones)[25-27]、α-酮醛肽类 (α-ketoaldehydes)[28]及乙烯砜肽类 (peptide vinyl sulfones)[29]等。其中, 二肽硼酸类似物硼替佐米 (Bortezomib, PS-341, Velcade®) 能够通过分子中硼酸部分与Thr1Oγ形成一个可逆的 (但解离速率十分缓慢) 四面体加合物并由于对蛋白酶体ChT-L活性抑制的高效力、高特异性和作用可逆性而被选择继续进行临床研究, 分别于2003年和2006年被FDA批准用于多发性骨髓瘤和套细胞淋巴瘤的治疗, 成为第一种作为化学治疗药物的PIs。但硼替佐米在治疗效力、安全性和给药方式上依然存在限制。具体而言, 临床研究中实体瘤抑制不理想, 推测至少部分与其在实体组织中的有限分布有关; 约1/3病患用药后出现周围神经病变这一高风险不良反应, 认为与其可以同时抑制对神经系统关键的丝氨酸蛋白酶有关; 需静脉注射, 造成一定不便。除此, 硼替佐米抗性逐渐出现[13, 15, 18]。目前, 不可逆作用的环氧酮肽类卡非佐米 (Carfilzomib, PR171, Kyprolis®) 和Oprozomib (ONX0912) 与硼酸肽类伊沙佐米 (Ixazomib, MLN9708, Ninlaro®) 和Delanzomib (CEP18770) 成为第二代PIs[19] (表 1)。卡非佐米是对一种放线菌属来源的名为epoxomicin的PI结构优化后获得的具有N-酰基吗啉帽的四肽α', β'-环氧酮分子。结构研究表明, 对于该类靶向分子, Thr1Oγ和Thr1N都会参与其α', β'-环氧酮的顺序双亲核攻击, 即Thr1Oγ首先攻击该药效团的羰基, 接着Thr1N打开环氧化物从而形成稳定的吗啉环加合物。考虑到N-末端氨基在该两步共价反应机制中的关键作用, α', β'-环氧酮衍生物对具有独特Thr1结构的蛋白酶体而非具有其他形式亲核基团的蛋白酶如丝氨酸蛋白酶、半胱氨酸蛋白酶表现出很高选择性。为得到效力更高、成药性更好的候选药物, epoxomicin的四肽部分被进行了优化。肽链长度研究表明四肽的选择性和效力最佳, 推测与蛋白酶体催化位点结合口袋大小有关; 四肽环氧酮的基本骨架确定后, 通过用不同氨基酸的不同基团修饰P2、P3、P4位置得到一系列epoxomicin衍生物并获得选择性和效力都很理想但水溶性低的YU-101; N-酰基吗啉帽的引入则导致具有更好成药性的卡非佐米的出现。与硼替佐米相比, 卡非佐米对蛋白酶体的CT-L活性抑制同样有效但更具选择性, 表现出外周神经病变的低发病率, 2012年7月被FDA批准用于已接受至少两种过往治疗的MM患者, 包括硼替佐米治疗和免疫调节治疗。但卡非佐米仍存在应用限制, 包括具有贫血、肺炎、血小板减少和中性粒细胞减少等不良反应, 同时共有硼替佐米频繁静脉给药等问题[25-27]。Ixazomib是FDA于2015年11月批准的与来那度胺和地塞米松联合用于已接受至少一种过往治疗的MM患者的首个口服PI。与硼替佐米相较, 同样优先靶向抑制β5亚基, 不存在交叉耐药, 有着改善的药代动力学和药效学参数[30]。Oprozomib和Delanzomib则分别是以卡非佐米和硼替佐米为先导化合物发展起来的具有改善的口服生物利用度的PI, 正处于Ⅰ/Ⅱ期临床试验以评价对恶性血液肿瘤和多种实体瘤的有效性和安全性[31]。
| Table 1 First and second generation proteasome inhibitors. MM: Multiple myeloma; MCL: Mantle cell lymphoma |
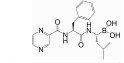
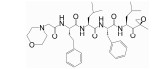
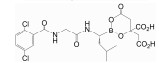
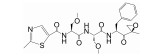
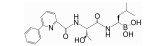
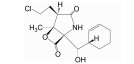
链霉菌属来源的aβ-内酯化合物 (aβ-lactones) 乳胞素 (lactacystin) 是首个发现的非肽类PI, 在体内转化为omuralide后通过分子中β-内酯不可逆共价修饰Thr1[32]。分离自海洋放线菌Salinispora tropica的一种omuralide类似物Marizomib (salinosporamide A, NPI-0052), 可以体外诱导Bortezomib耐药MM细胞凋亡并延长荷瘤动物的存活时间, 由于作用效力高于硼替佐米且具有良好口服生物利用度, 针对晚期实体瘤、淋巴瘤及MM的Ⅰ/Ⅱ期临床试验正在进行[33, 34]。
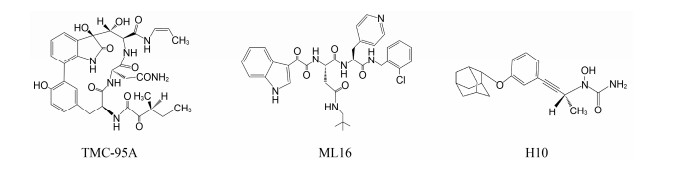
2.1.2 非共价作用型共价作用的CP靶向PIs在临床上具有的高风险不良反应和不断出现的耐药性以及在多种实体瘤动物模型中表现出的低效力表明了开发具有不同作用机制PIs的必要性。非共价作用的CP靶向分子由于不存在易被亲核攻击的反应活性基团, 对蛋白酶体作用表现出较高的选择性和较低的过度反应性及不稳定性, 因而低毒且不良反应少。此外, 快速结合和分离的动力学特点允许药物在体内有着更广泛的组织分布, 包括液体瘤和实体瘤组织[35]。目前, 对某些肽类、拟肽类和几种有机化合物作为非共价作用CP靶向抑制剂的研究有了初步进展。其中, 已获得足够结构信息支持非共价作用机制的有天然环肽类TMC-95A及其合成衍生物、合成拟肽类ML16及其衍生物和羟基脲衍生物H10[35, 36]。分离自真菌蒙塔涅梨孢假壳 (Apiospora montagnei) 的TMC-95系列化合物是首个发现的非共价作用的β折叠样底物模拟物类PI, 分子中有一个大环并通过形成反向β折叠样氢键网络结合至β亚基。其中, TMC-95A具有的芳香族大环能够与β亚基S2和S4口袋很好契合, 而大部分其他蛋白酶无法容纳空间上要求苛刻的大环[37], 因而表现出对蛋白酶体作用的高效性和高选择性。TMC-95系列的发现为发展非共价作用CP靶向分子提供了基础。从千年制药公司 (Millennium Pharmaceuticals) 一个化合物库中筛选获得并经晶体结构研究确证了一种非共价作用CP靶向抑制剂——具有S-高苯丙氨酸的N, C-加帽三肽, 结构优化后获得的系列二肽化合物之一ML16, 其P3处新戊基-天冬酰胺基团能够对S3进行优化填充, 不仅具有对蛋白酶体作用的高效性和高选择性, 还具有良好的胞内活性[38, 39]。H10是在一个经体外筛选程序鉴定出的作为CP靶向抑制剂的含羟基脲化合物基础上设计、合成的非肽类羟基脲衍生物, 晶体结构研究揭示其金刚烷基和苯基能够优化填充β亚基S3和S1。羟基脲化合物的药代动力学特性将为PIs开发开辟新途径[40]。
2.2 iCP选择性靶向抑制剂与传统CP靶向抑制相比, 选择性靶向抑制iCP在抗骨髓瘤和抗淋巴瘤活性基本保持不变的同时治疗指数显著增加。iCP选择性靶向抑制剂不仅对恶性血液病, 对炎症和自身免疫性疾病也代表了一种更具前景的治疗策略[41]。小鼠iCP和cCP晶体结构研究表明, 与相应βc相比, β5i底物结合凹槽氨基酸组成和排列变化微小因而ChT-L活性不变, 但具有更为宽敞的S1结合口袋和更为有限且亲水性增强的S3结构口袋; β1i底物结合通道疏水氨基酸的排列使其C-L活性大大降低而表现ChT-L活性; β2i与β2c结构相似, 仅Asp53 (β2c) 被Glu (β2i) 取代, 仍表现T-L活性[42]。因此, β5i选择性抑制剂需要靶向分子P1位点有大的疏水氨基酸和P3有小的极性氨基酸, β1i选择性抑制剂需要P1有分支的非极性氨基酸和P3有小的极性氨基酸[43]。环氧酮肽类ONX0914/PR-957, 由于其P1苯基侧链能够与β5i宽敞的S1口袋很好契合却不能和β5c作用而成为有效的β5i选择性抑制剂。它可以减少激活的单核细胞和T细胞细胞因子的生成, 减缓小鼠模型中类风湿性关节炎、全身性红斑狼疮、结肠炎及脑脊髓炎等疾病进程[44, 45]。Lin等[46, 47]观察到结核分枝杆菌 (Mycobacterium tuberculosis, Mtb) 20S蛋白酶体β亚基和免疫蛋白酶体β5i在S1口袋处均偏好大的芳香残基, 通过对Mtb 20S蛋白酶体选择性靶向的非共价作用N, C-加帽二肽进行结构优化开发出系列基于β-氨基酸的拟肽类iCP选择性靶向抑制剂。其中, 高效力的PKS3054表现出对人T细胞激活的抑制并未有明显毒性。肽醛类IPSI-001对β1i和β5i尤其是β1i具有选择性抑制作用, 可以诱导MM细胞及其他恶性血液病细胞的体外凋亡[48]。总之, βi和βc活性位点间的所有结构差异都将进一步被开发利用以增强对cCP/iCP的选择靶向性, 同时, βi活性位点的结构信息也将指导设计具有更高亲和力的iCP靶向分子。
|
真核蛋白酶体各类RP在底物降解之前执行系列准备和协助工作, 干扰RP作用可以抑制蛋白酶体功能并部分克服MM对CP靶向分子不断出现的耐药性, 代表了一种有潜力的治疗策略。目前鉴定出3类19S相关分子可以作为药物作用靶标: AAA ATP酶 (ATPases associated with diverse cellular activities, AAA/AAA+)、泛素受体 (ubiquitin receptors) 和去泛素化酶 (deubiquitylating enzymes, DUBs)。其中, 对19S DUBs靶向抑制剂的研究相对较广泛。尽管19S靶向分子的开发落后于CP靶向分子, 近期关于19S亚复合体结构研究的进展将促进该类靶向抑制剂的开发[8, 9]。
哺乳动物19S DUBs包括JAMM家族的POH1 (也称作RPN11)、UCHs家族的UCHL5 (也称作UCH37) 和USP家族的USP14。稳定整合入19S的Zn2+依赖金属蛋白酶RPN11在底物与Rpt环结合后移除底物泛素链以促进其易位。已知癌细胞中的过表达能够削弱细胞毒试剂的作用且Rpn11和EGFR联合抑制表现出对小细胞肺癌治疗的益处[49]。近期对Rpn11-Rpn8这一蛋白酶体去泛素化酶二聚体结构的鉴定将为发展Rpn11靶向抑制剂提供一定基础[50]。与19S可逆互作的半胱氨酸异肽酶UCHL5和USP14从远端对泛素链进行解组装, 对这一并不与底物降解直接偶联的泛素链修剪过程的确切生理角色还不清楚, 但抑制两者之一会上调蛋白酶体活性而联合抑制阻断蛋白酶体功能并致MM细胞凋亡[51, 52]。D’Arcy等[51, 53]表明小分子b-AP1能够同时靶向并可逆作用于USP14和UCHL5, 从而诱导Bortezomib耐药MM细胞凋亡并在几种实体瘤模型中表现出抗癌活性, 其衍生物VLX1570由于更高的效力和良好的水溶性处于Ⅰ/Ⅱ期临床试验阶段以评价对复发性/难治性MM患者的安全性和有效性[54, 55]。
4 结语与展望目前, 蛋白酶体20S CP靶向的药物开发在晶体结构研究的指导下已得到极大推进, 尤其是CP变种的晶体结构研究促使选择性靶向分子包括促进机制研究的特异工具化合物、针对自身免疫疾病的治疗试剂和抗病原体候选物的设计得以实现。相对的, 由于26S蛋白酶体制备物构象和组成上的异质性使得适于X-射线晶体衍射的全酶晶体获得较晚, 19S靶向抑制剂的开发还未取得明显成效, 相信随着对19S结构、作用机制和生理角色理解的深入, 更多靶向分子会得以开发。需要注意的是, PIs具有难以避免的临床应用限制, 包括由于广泛的细胞影响和多种作用底物产生的高风险不良反应及不断出现的抗药性。已有研究者将注意力转向靶向UPS的上游事件, 即E1利用ATP激活泛素并转移给E2, E3结合并促进泛素转移至其底物从而引导26S蛋白酶体对泛素化底物的降解[56]。很明显, E3泛素连接酶对将要被泛素化和进一步降解的特异性底物的确定至关重要。相对于靶向UPS下游事件的26S蛋白酶体抑制策略, E3连接酶抑制可以靶向特异的蛋白质或信号转导途径从而减少PIs在体内普遍的脱靶效应, 有望成为肿瘤治疗的新型策略。目前, 几种新化合物已在临床/临床前研究中表现出积极效果[57, 58]。
| [1] | Glickman MH, Rubin DM, Fried VA, et al. The regulatory particle of the Saccharomyces cerevisiae proteasome[J]. Mol Cell Biol, 1998, 18: 3149–3162. DOI:10.1128/MCB.18.6.3149 |
| [2] | Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome[J]. Annu Rev Biochem, 2009, 78: 477–513. DOI:10.1146/annurev.biochem.78.081507.101607 |
| [3] | Ciechanover A. Intracellular protein degradation:from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting[J]. Bioorg Med Chem, 2013, 21: 3400–3410. DOI:10.1016/j.bmc.2013.01.056 |
| [4] | Groll M, Ditzel L, Lowe J, et al. Structure of 20S proteasome from yeast at 2.4Å resolution[J]. Nature, 1997, 386: 463–471. DOI:10.1038/386463a0 |
| [5] | Kisselev AF, Callard A, Goldberg AL. Importance of different active sites in protein breakdown by 26S proteasomes and the efficacy of proteasome inhibitors varies with the protein substrate[J]. J Biol Chem, 2006, 281: 8583–8590. |
| [6] | Groll M, Bajorek M, Köhler A, et al. A gated channel into the proteasome core particle[J]. Nat Struct Biol, 2000, 7: 1062–1067. DOI:10.1038/80992 |
| [7] | Stadtmeller BM, Hill CP. Proteasome activators[J]. Mol Cell, 2011, 41: 8–19. DOI:10.1016/j.molcel.2010.12.020 |
| [8] | Lander GC, Estrin E, Matyskiela ME, et al. Complete subunit architecture of the proteasome regulatory particle[J]. Nature, 2012, 482: 186–191. |
| [9] | Lasker K, Forster F, Bohn S, et al. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach[J]. Proc Natl Acad Sci U S A, 2012, 109: 1380–1387. DOI:10.1073/pnas.1120559109 |
| [10] | Kloetzel PM. Antigen processing by the proteasome[J]. Nat Rev Mol Cell Biol, 2001, 2: 179–187. DOI:10.1038/35056572 |
| [11] | Kincaid EZ, Che JW, York I, et al. Mice completely lacking immunoproteasomes show major changes in antigen presentation[J]. Nat Immunol, 2012, 13: 129–135. |
| [12] | King RW, Deshaies RJ, Peters JM, et al. How proteolysis drives the cell cycle[J]. Science, 1996, 274: 1652–1659. DOI:10.1126/science.274.5293.1652 |
| [13] | Adams J. The development of proteasome inhibitors as anticancer drugs[J]. Cancer Cell, 2004, 5: 417–421. DOI:10.1016/S1535-6108(04)00120-5 |
| [14] | Chen L, Madura K. Increased proteasome activity, ubiq-uitin-conjugating enzymes, and eEF1A translation factor detected in breast cancer tissue[J]. Cancer Res, 2005, 65: 5599–5606. DOI:10.1158/0008-5472.CAN-05-0201 |
| [15] | Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome inhibitors:an expanding army attacking a unique target[J]. Chem Biol, 2012, 19: 99–115. DOI:10.1016/j.chembiol.2012.01.003 |
| [16] | Teicher BA, Tomaszewski JE. Proteasome inhibitors[J]. Biochem Pharmacol, 2015, 96: 1–9. DOI:10.1016/j.bcp.2015.04.008 |
| [17] | Verbrugge SE, Scheper RJ, Lems WF, et al. Proteasome inhibitors as experimental therapeutics of autoimmune dis-eases[J]. Arthritis Res Ther, 2015, 17: 17. DOI:10.1186/s13075-015-0529-1 |
| [18] | Groll M, Huber R, Moroder L. The persisting challenge of selective and specific proteasome inhibition[J]. J Pept Sci, 2009, 15: 58–66. DOI:10.1002/psc.v15:2 |
| [19] | Ettari R, Previti S, Bitto A, et al. Immunoprotea-some-selective inhibitors:a promising strategy to treat hematologic malignancies, autoimmune and inflammatory diseases[J]. Curr Med Chem, 2016, 23: 1217–1238. DOI:10.2174/0929867323666160318173706 |
| [20] | Li H, O'Donoghue AJ, van der Linden WA, et al. Structure-and function-based design of plasmodium-selective proteasome inhibitors[J]. Nature, 2016, 530: 233–236. DOI:10.1038/nature16936 |
| [21] | Borissenko L, Groll M. 20S proteasome and its inhibitors:crystallographic knowledge for drug development[J]. Chem Rev, 2007, 107: 687–717. DOI:10.1021/cr0502504 |
| [22] | Kisselev AF, van der Linden WA, Overkleeft HS. Pro-teasome inhibitors:an expanding army attacking a unique target[J]. Chem Biol, 2012, 19: 99–115. DOI:10.1016/j.chembiol.2012.01.003 |
| [23] | Lee DH, Goldberg AL. Selective inhibitors of the protea-some-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae[J]. J Biol Chem, 1996, 271: 27280–27284. DOI:10.1074/jbc.271.44.27280 |
| [24] | Braun HA, Umbreen S, Groll M, et al. Tripeptide mimetics inhibit the 20S proteasome by covalent bonding to the active threonines[J]. J Biol Chem, 2005, 280: 28394–28401. DOI:10.1074/jbc.M502453200 |
| [25] | Kim KB, Crews CM. From epoxomicin to carfilzomib:chemistry, biology, and medical outcomes[J]. Nat Prod Rep, 2013, 30: 600–604. DOI:10.1039/c3np20126k |
| [26] | Zhang J, Wu P, Hu Y. Clinical and marketed proteasome inhibitors for cancer treatment[J]. Curr Med Chem, 2013, 20: 2537–2551. DOI:10.2174/09298673113209990122 |
| [27] | Perel G, Bliss J, Thomas CM. Carfilzomib (Kyprolis):a novel proteasome inhibitor for relapsed and/or refractory multiple myeloma[J]. Pharm Ther, 2016, 41: 303–307. |
| [28] | Grawert MA, Gallastegui N, Stein M, et al. Elucidation of the α-keto-aldehyde binding mechanism:a lead structure motif for proteasome inhibition[J]. Angew Chem Int Ed Engl, 2011, 50: 542–544. DOI:10.1002/anie.201005488 |
| [29] | Geurink PP, van der Linden WA, Mirabella AC, et al. Incorporation of non-natural amino acids improves cell permeability and potency of specific inhibitors of proteasome trypsin-like sites[J]. J Med Chem, 2013, 56: 1262–1275. DOI:10.1021/jm3016987 |
| [30] | Shirley M. Ixazomib:first global approval[J]. Drugs, 2016, 76: 405–411. DOI:10.1007/s40265-016-0548-5 |
| [31] | Kroiss M, Sbiera S, Kendl S, et al. Drug synergism of proteasome inhibitors and mitotane by complementary activetion of ER stress in adrenocortical carcinoma cells[J]. Horm Cancer, 2016, 7: 345–355. DOI:10.1007/s12672-016-0273-2 |
| [32] | Dick LR, Cruikshank AA, Destree AT, et al. Mechanistic studies on the inactivation of the proteasome by lactacystin in cultured cells[J]. J Biol Chem, 1997, 272: 182–188. DOI:10.1074/jbc.272.1.182 |
| [33] | Groll M, McArthur KA, Macherla VR, et al. Snapshots of the fluorosalinosporamide/20S complex offer mechanistic insights for fine tuning proteasome inhibition[J]. J Med Chem, 2009, 52: 5420–5428. DOI:10.1021/jm900559x |
| [34] | Harrison SJ, Mainwaring P, Price T, et al. Phase Ⅰ clinical trial of marizomib (NPI-0052) in patients with advanced malignancies including multiple myeloma:study NPI-0052-102 final results[J]. Clin Cancer Res, 2016, 22: 4559–4566. DOI:10.1158/1078-0432.CCR-15-2616 |
| [35] | Kaffy J, Bernadat G, Ongeri S. Non-covalent proteasome inhibitors[J]. Curr Pharm Des, 2013, 19: 4115–4130. DOI:10.2174/1381612811319220016 |
| [36] | Di Giovanni C, Ettari R, Sarno S, et al. Identification of noncovalent proteasome inhibitors with high selectivity for chymotrypsin-like activity by a multistep structure-based virtual screening[J]. Eur J Med Chem, 2016, 121: 578–591. DOI:10.1016/j.ejmech.2016.05.049 |
| [37] | Kaiser M, Groll M, Siciliano C, et al. Binding mode of TMC-95A analogues to eukaryotic 20S proteasome[J]. ChemBioChem, 2004, 5: 1256–1266. DOI:10.1002/cbic.v5:9 |
| [38] | Blackburn C, Gigstad KM, Hales P, et al. Characterization of a new series of non-covalent proteasome inhibitors with exquisite potency and selectivity for the 20S beta5-subunit[J]. Biochem J, 2010, 430: 461–476. DOI:10.1042/BJ20100383 |
| [39] | Blackburn C, Barrett C, Blank JL, et al. Optimization of a series of dipeptides with a P3 threonine residue as non-covalent inhibitors of the chymotrypsin-like activity of the human 20S proteasome[J]. Bioorg Med Chem Lett, 2010, 20: 6581–6586. DOI:10.1016/j.bmcl.2010.09.032 |
| [40] | Gallastegui N, Beck P, Arciniega M, et al. Hydroxyureas as noncovalent proteasome inhibitors[J]. Angew Chem Int Ed Engl, 2012, 51: 247–249. DOI:10.1002/anie.201106010 |
| [41] | Ettari R, Previti S, Bitto A, et al. Immunoproteasome-selective inhibitors:a promising strategy to treat hematologic malignancies, autoimmune and inflammatory diseases[J]. Curr Med Chem, 2016, 23: 1217–1238. DOI:10.2174/0929867323666160318173706 |
| [42] | Huber EM, Basler M, Schwab R, et al. Immuno-and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity[J]. Cell, 2012, 148: 727–738. DOI:10.1016/j.cell.2011.12.030 |
| [43] | Kasam V, Lee NR, Kim KB, et al. Selective immunoproteasome inhibitors with non-peptide scaffolds identified from structure-based virtual screening[J]. Bioorg Med Chem Lett, 2014, 24: 3614–3617. DOI:10.1016/j.bmcl.2014.05.025 |
| [44] | Muchamuel T, Basler M, Aujay MA, et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis[J]. Nat Med, 2009, 15: 781–787. DOI:10.1038/nm.1978 |
| [45] | Mishto M, Raza ML, de Biase D, et al. The immunoproteasome β5i subunit is a key contributor to ictogenesis in a rat model of chronic epilepsy[J]. Brain Behav Immun, 2015, 49: 188–196. DOI:10.1016/j.bbi.2015.05.007 |
| [46] | Lin G, Chidawanyika T, Tsu C, et al. N, C-capped dipeptides with selectivity for mycobacterial proteasome over human proteasomes:role of S3 and S1 binding pockets[J]. J Am Chem Soc, 2013, 135: 9968–9971. DOI:10.1021/ja400021x |
| [47] | Singh PK, Fan H, Jiang X, et al. Immunoproteasome β5i-selective dipeptidomimetic inhibitors[J]. ChemMedChem, 2016, 11: 2127–2131. DOI:10.1002/cmdc.v11.19 |
| [48] | Kuhn DJ, Hunsucker SA, Chen Q, et al. Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors[J]. Blood, 2009, 113: 4667–4676. DOI:10.1182/blood-2008-07-171637 |
| [49] | Bivona TG, Hieronymus H, Parker J, et al. FAS and NF-κB signaling modulate dependence of lung cancers on mutant EGFR[J]. Nature, 2011, 471: 523–526. DOI:10.1038/nature09870 |
| [50] | Pathare GR, Nagy I, Sledz P, et al. Crystal structure of the proteasomal deubiquitylation module Rpn8-Rpn11[J]. Proc Natl Acad Sci U S A, 2014, 111: 2984–2989. DOI:10.1073/pnas.1400546111 |
| [51] | D'Arcy P, Linder S. Proteasome deubiquitinases as novel targets for cancer therapy[J]. Int J Biochem Cell Biol, 2012, 44: 1729–1738. DOI:10.1016/j.biocel.2012.07.011 |
| [52] | Liu CW, Jacobson AD. Functions of the 19S complex in proteasomal degradation[J]. Trends Biochem Sci, 2013, 38: 103–110. DOI:10.1016/j.tibs.2012.11.009 |
| [53] | Tian Z, D'Arcy P, Wang X, et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance[J]. Blood, 2014, 123: 706–716. DOI:10.1182/blood-2013-05-500033 |
| [54] | Wang X, D'Arcy P, Caulfield TR, et al. Synthesis and evaluation of derivatives of the proteasome deubiquitinase inhibitor b-AP15[J]. Chem Biol Drug, 2015, 86: 1036–1048. DOI:10.1111/cbdd.12571 |
| [55] | Wang X, Mazurkiewicz M, Hillert EK, et al. The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells[J]. Sci Rep, 2016, 6: 26979. DOI:10.1038/srep26979 |
| [56] | Liu J, Shaik S, Dai X, et al. Targeting the ubiquitin pathway for cancer treatment[J]. Biochim Biophys Acta, 2015, 1855: 50–60. |
| [57] | Liu Y, Mallampalli RK. Small molecule therapeutics targeting F-box proteins in cancer[J]. Semin Cancer Biol, 2016, 36: 105–119. DOI:10.1016/j.semcancer.2015.09.014 |
| [58] | Shah JJ, Jakubowiak AJ, O'Connor OA, et al. Phase Ⅰ study of the novel investigational NEDD8-activating enzyme inhibitor pevonedistat (MLN4924) in patients with relapsed/refractory multiple myeloma or lymphoma[J]. Clin Cancer Res, 2016, 22: 34–43. |