 2016, Vol. 51
2016, Vol. 51



2. 中南民族大学药学院, 湖北 武汉 430071
2. College of Pharmacy, South Central University for Nationalities, Wuhan 430071, China
组蛋白是真核生物染色体的基本组成单位, 组蛋白的乙酰化和去乙酰化是基因表达过程中的重要调控方式, 是转录过程中的关键修饰[1, 2]。在正常生理状态下, 组蛋白的乙酰化和去乙酰化是一个动态平衡过程, 由组蛋白乙酰化转移酶(histone acetyl-transferases, HATs)和组蛋白去乙酰化酶(histone deacetylases, HDACs)协调控制[3, 4]。当组蛋白去乙酰化水平增加, 乙酰化水平相对降低, 使得原有的基因表达平衡状态被打破, 导致正常细胞周期与代谢行为改变并诱发肿瘤, 尤其是HDAC1、HDAC2、HDAC6在多种肿瘤细胞中过表达[5-7]。因此, HDACs已成为近年来一个新兴的抗肿瘤靶点, 而组蛋白去乙酰化酶抑制剂(histone deacetylase inhibitor, HDACi)也成为国内外抗肿瘤药物的研究热点[8, 9]。自第一个组蛋白去乙酰化酶抑制剂Vorinostat (SAHA)被美国FDA批准用于治疗皮肤T细胞淋巴癌后[10], 目前已有数百个HDACi被报道了。
癌症是一个高度复杂、多基因调控的疾病, 涉及到多种交叉信号通路。采用单一靶点药物来治疗这种复杂的疾病, 通常难以达到理想的效果, 甚至会出现耐药性和严重的不良反应。因此, 设计出可以同时作用于多个靶点的药物, 已经成为当今药物研发的热点和难点。HDACi通常是由表面识别区域、脂肪链和金属锌离子结合基团3部分组成, 当HDACi与HDACs相互作用后, HDACi的表面识别区域仅仅占据活性口袋入口处的一小部分[3]。因此, 将HDACi表面识别区域拼接另一抗肿瘤药物不仅有利于增强整个抑制剂与HDACs的结合作用, 提高其抗癌活性, 而且设计出的分子还具有双靶向性。基于这个原理, 目前已经报道了多个HDACi的双靶点分子, 例如将雌激素配体乙炔雌二醇和他莫昔芬与HDACi (SAHA)拼接得到的缀合物(化合物1A和1B, 图 1)就是一类双靶点药物, 比单一使用他莫昔芬或SAHA有更强的抗乳腺癌的效果[11]。
 | Figure 1 Dual-acting ER ligands-HDACi conjugates |
近年来研究表明, 喹诺酮类抗菌剂在细菌内的作用靶点拓扑异构酶Ⅱ与哺乳动物的拓扑异构酶Ⅱ在活性部分酪氨酸周围的序列具有同源性, 而哺乳动物拓扑异构酶Ⅱ已经是某些抗肿瘤药物的靶点如多柔比星; 此外, 喹诺酮类药物抗肿瘤机制和杀菌的机制相似, 都是通过抑制拓扑异构酶Ⅱ和DNA形成的复合物[12], 这使得喹诺酮类药物有望被开发成一类新型结构的抗肿瘤药物。为了进一步增强喹诺酮类药物的抗肿瘤活性或改善其理化性质, 人们对其展开了大量的结构修饰及构效关系研究[13-15]。本文也以环丙沙星为先导化合物并对其进行结构修饰, 拟将HDACi (SAHA)引入到环丙沙星来构建环丙沙星-HDACi缀合物, 以期得到新型、高效的环丙沙星衍生物。
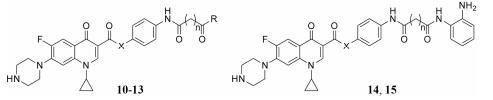
参考喹诺酮类化合物结构改造经验发现, 喹诺酮类药物C3位羧基被其他基团取代能够提高抗肿瘤活性[16, 17]。因此, 本研究将HDACi (SAHA)引入到环丙沙星的C3位(化合物12、13); 同时, 为了进一步研究缀合物的构效关系, 羧酸类化合物(化合物10、11)和2-苯酰胺类化合物(化合物14、15)也被合成了。测试了目标化合物对HDACs的抑制活性和体外抗癌活性。结果表明, 目标化合物对HDAC1、HDAC2和HDAC6均有较强的抑制活性, 尤其是缀合物12b对HDAC1和HDAC6的抑制活性强于阳性药物SAHA; 在体外抗肿瘤活性中, 目标化合物对人大细胞肺癌NCI-H460、乳腺癌MCF-7、肺癌A549、人口腔上皮样癌KB和人宫颈癌HeLa细胞5种肿瘤细胞均有抑制活性, 并强于环丙沙星。其中缀合物12b对NCI-H460和A549也展现出了最强的抑制活性, 分别是阳性药物SAHA的7倍和5.7倍。所合成的18个新化合物均未见文献报道。
结果与讨论 1 目标化合物的合成目标化合物10~15的合成过程见合成路线1。首先, 将二酸类化合物1与醋酸酐反应转化成相应的酸酐2; 接着与取代苯胺反应制得酰胺类化合物5和6; 随后用三氟乙酸将化合物5的Boc脱去、BBr3将化合物6的甲基脱去, 分别得到氨基化合物7和羟基化合物8; 然后将化合物7、8在缩合剂的作用下直接与环丙沙星9进行偶联得到了化合物10、11, 再用羟胺将化合物10、11的羧酸转化为异羟肟酸得到了相应的目标化合物12、13。另一方面, 将羧酸化合物10、11直接与邻苯二胺反应制得2-苯酰胺类目标化合物14、15。目标化合物的结构经1H NMR和HR-MS分析确证, 其收率、理化常数及波谱数据见表 1、2。
| Table 1 Physical contants and HR-MS of synthesized compounds 10-15 |
| Table 2 The 1H NMR data of target compounds (400 MHz, acetone-d6) |

 | Scheme1 Synthetic route of ciprofloxacin-HDACi conjugates 10-15. Reagents and conditions: (a) Ac2O, THF, reflux, 1 h; (b) THF, rt, 3 h; (c) TFA, CH2Cl2, 0 ℃, 1 h; (d) BBr3, CH2Cl2, -20 ℃, 12 h; (e) EDC, HOBT, CH2Cl2, rt, 24 h for 12; EDC, DMAP, CH2Cl2, rt, 24 h for 13; (f) THF, 0 ℃, Ar, 15 min and then NH2OH·HCl, KOH, MeOH, rt, 1 h; (g) (i) (C2H5)3N, BOP reagent, dry DMF, 0.5 h, rt; (ii) o-Phenylendiamine, dry DMF, rt, 12 h |
目标化合物对HDACs的抑制活性结果见表 3。从表 3可以发现, 所合成的18个环丙沙星-HDACi缀合物对HDAC1、HDAC2和HDAC6均有较强的抑制活性, 尤其是肟酸(化合物12、13)和2-苯酰胺(化合物14、15)系列化合物对HDAC1、HDAC2和HDAC6的抑制活性均在纳摩尔水平。这些结果说明目标缀合物具有较强的HDAC靶向性。从结构上看, 合成的缀合物是用环丙沙星取代了SAHA的苯环, 在HDAC活性口袋中SAHA的苯环仅占据了HDAC活性口袋入口处的少量空间。因此, 将环丙沙星引入到SAHA中能够增强化合物与HDAC的相互作用, 从而使得环丙沙星-HDACi缀合物也有较强的HDAC抑制活性。但是, 目标缀合物HDACi单元的侧链长度对活性影响较大, 其中以6个亚甲基(n=6)连接的化合物活性最佳, 延长(n=7)或缩短(n=5)均减弱了HDACs的抑制活性, 例如在化合物10系列中, 化合物10b (n=6)对HDACs的抑制活性强于化合物10a (n=5)和化合物10c (n=7)。此外, 目标化合物中的Zn离子结合基团(羧基、肟酸基和2-苯酰胺基)对HDACs的抑制活性也有较大的影响, 其中肟酸基团(化合物12、13)对HDAC1、HDAC2和HDAC6的抑制活性明显强于相应的羧基(化合物10、11)和2-苯酰胺基(化合物14、15)。特别是肟酸类化合物12b在所有化合物中对HDAC1、HDAC2和HDAC6的抑制活性最强, 其IC50分别为0.041、0.042、0.039 μmol·L-1, 对HDAC1和HDAC6的抑制活性甚至略强于阳性药物SAHA, 这可能是由于HDAC1、HDAC2和HDAC6是一个Zn离子依赖型的酶, 而肟酸基团比羧酸和2-苯酰胺基与Zn离子有更强的相互作用力, 使得肟酸类化合物对HDACs的抑制活性强于羧基和2-苯酰胺基类化合物。环丙沙星与HDACi的连接基团对酶的抑制活性也有较大的影响, 其中酰胺连接键的活性明显高于酯键, 例如将化合物12b连接链由酰胺键换成酯键(化合物13b)后对HDAC1、HDAC2和HDAC6的抑制活性显著降低。
| Table 3 In vitro inhibition of HDAC1, HDAC2 and HDAC6 of target compounds. SAHA: Vorinostat |
通过HDACs抑制实验发现肟酸化合物12b对HDAC1和HDAC6展现出了最强的抑制活性, 通过Autodock4.2D分子模拟软件将化合物12b与HDAC1和HDAC6进行了分子对接, 对接结果如图 2所示。在HDAC1和HDAC6的口袋中, 化合物12b的表面识别区环丙沙星单元均位于活性口袋的入口处(图 2A、B), 并与SAHA的表面识别区苯环相比占据了更多的空间, 因此引入环丙沙星单元增强了SAHA与酶的相互作用。此外, HDACi的表面识别区与酶活性边缘氨基酸残基相互作用, 不仅影响对酶的抑制效果, 还将决定HDACi对酶亚型的选择性[18]。在HDAC1中, 化合物12b喹诺酮母核上的N原子能够与酶氨基酸残基G27形成氢键(图 2C), 而在HDAC6中该N原子没有形成氢键, 但喹诺酮母核上的羰基却与S568形成了氢键(图 2D), 因此化合物12b对HDAC1和HDC6有明显的选择性和抑制活性。另外, 化合物12b中SAHA单元的脂肪链与酶活性口袋的通道壁形成了较强的疏水作用(图 2A、B), 肟酸基团与活性口袋底部的氨基酸残基相互作用的同时还能够与活性口袋底部的Zn2+形成配位, 因此化合物12b对HDAC1和HDAC6均有较强的抑制活性。但是化合物12b的肟酸基团在HDAC1和HDAC6活性口袋底部与氨基酸残基和锌离子作用方式不同。在HDAC1中, 肟酸基团与H141、D176、H178、D264和Y303氨基酸残基相互作用形成了5个氢键, 并且肟酸基团的羟基(3.1Å)和羰基(2.7Å)能够与氨基酸残基D176 (2.3Å)、D178 (2.0Å)、D264 (1.9Å)一起与Zn2+形成六配位(图 2C); 而在HDAC6中, 化合物12b的肟酸基团与D649、H651和Y782氨基酸残基相互作用仅形成了3个氢键, 同时肟酸基团的羟基氧(3.3Å)和羰基氧(2.5Å)能够与氨基酸残基D649 (2.1Å)、H651 (1.9Å)、D742 (2.7Å)一起与Zn2+形成五配位(图 2D)。化合物12b的SAHA单元除了与活性口袋底部的氨基酸残基和锌离相互作用外, 化合物12b在HDAC6中, 其SAHA单元的酰胺连接基团的羰基还能与氨基酸残基N654形成氢键作用, 而在HDAC1中酰胺连接基团并没有形成氢键, 因此化合物12b对HDAC6的抑制活性要强于HDAC1。
 | Figure 2 The predicted binding modes of compound 12b-HDAC1 and HDAC6. (A) Molecular surface of the HDAC1 binding pocket with docked compound 12b. (B) Molecular surface of the HDAC6 binding pocket with docked compound 12b. (C) Docking poses of HDAC1-12b, which can form hydrogen bonds with residues G27, H141, D176, H178, D264, Y303, and which can coordinate the zinc ion with residues D176, H178, D264. (D) Docking poses of HDAC6-12b, which can form hydrogen bonds with residues S568, D649, H651, N654, Y782, and which can coordinate the zinc ion with residues D649, H651, D742. Distances are given in Å |
从体外抑制肿瘤活性实验结果表 4可见, 这些环丙沙星-HDACi缀合物对NCI-H460、MCF-7、A549、KB和HeLa 5种肿瘤细胞均有不同程度的抑制作用, 其IC50值均小于32.0 μmol·L-1, 显著低于先导化合物环丙沙星的IC50 ( > 65 μmol·L-1)值。缀合物对人大细胞肺癌NCI-H460细胞展现出了更强的抑制活性, 如化合物12a~12c和14b~14c对NCI-H460的抑制活性甚至强于阳性药物SAHA, 其中化合物12b和14b的IC50达到了纳摩尔水平。这些实验结果表明引入HDACi能够增强环丙沙星的抗肿瘤活性。在HDACs抑制活性中, 目标缀合物的HDACi单元侧链长度对活性有较大影响, 以6个亚甲基(n=6)连接的化合物活性最佳; 同样的, 在抗肿瘤活性中也有着类似的趋势即6个亚甲基(n=6)连接的化合物抗肿瘤活性最强, 延长或缩短链长均降低抗肿瘤活性, 例如化合物12b (n=6)对NCI-H460的IC50为0.7 μmol·L-1, 将其侧链缩短(n=5, 化合物12a, IC50=2.3 μmol·L-1)或延长(n=7, 化合物12c, IC50=1.2 μmol·L-1)后均降低了抗肿瘤活性。此外, 与HDACs的抑制活性一致, 肟酸化合物的抗肿瘤活性强于羧酸和2-苯酰胺基化合物, 如表 4所示。特别是肟酸化合物12b对NCI-H460和A549均展现出了最强的抑制活性, 分别是阳性药物SAHA的7倍和5.7倍。但是, 将肟酸类缀合物中环丙沙星与HDACi单元的连接基团由酰胺键转化成酯键后显著地降低了抗肿瘤活性。事实上, 相对酰胺类化合物而言, 酯类化合物对5种肿瘤细胞仅展现出了中等的抑制活性(IC50 > 15 μmol·L-1)。
| Table 4 Whole cell antiproliferative activity of target compounds (n=3) |
另外, 为了进一步考察目标化合物对肿瘤细胞的选择性以及对正常细胞的毒性, 用正常肺上皮细胞BEAS-2B和正常乳腺上皮细胞MCF-10A作为对照细胞来检测HDACs和对人大细胞肺癌NCI-H460、乳腺癌MCF-7、肺癌A549均有较强抑制活性的化合物12b~12c和14b~14c的抗肿瘤选择性以及毒性, 结果如表 5所示。缀合物对NCI-H460、MCF-7、A549三种肿瘤细胞的抑制活性(IC50均小于8 μmol·L-1)结果表明, 化合物12c和14b~14c对正常的BEAS-2B细胞或正常的MCF-10A细胞仅展现出了中等抑制活性(IC50均大于38 μmol·L-1), 化合物12b对这两种正常细胞均没有抑制活性, 说明缀合物对肿瘤细胞有较强选择性, 并对正常的细胞的毒性较低, 其中化合物12b没有毒性, 而阳性药物SAHA对两种正常细胞均表现出了较强的毒性。
| Table 5 Anticancer activity of compounds 12b-12c and 14b-14c (n=3) |
本研究基于HDACi表现出的优秀抗肿瘤活性, 通过酯化或酰胺化反应将HDACi引入到环丙沙星中, 合成了18个环丙沙星-HDACi缀合物。经测试目标化合物对HDACs的抑制活性发现, 目标化合物对HDAC1、HDAC2和HDAC6均有较强的抑制活性, 但是HDACi单元的链长、锌离子结合基团以及环丙沙星与HDACi的连接基团对活性均有较大的影响, 以链长为6个亚甲基(n=6)、锌离子的结合基团为肟酸、连接基团为酰胺键的化合物具有较强的HDACs抑制活性。其中化合物12b展现出了最强的HDACs抑制活性, 其对HDAC1和HDAC6的抑制活性甚至略强于阳性药物SAHA。体外抗瘤活性发现, 目标化合物对人大细胞肺癌NCI-H460、乳腺癌MCF-7、肺癌A549、人口腔上皮样癌KB和人宫颈癌HeLa细胞均有抑制活性。尤其是对人大细胞肺癌NCI-H460有较强的抑制活性和选择性, 并且化合物抗癌活性和酶抑制活性的趋势一致, 其中化合物12b对NCI-H460和A549也有最强的抑制活性, 并强于先导化合物环丙沙星和SAHA。此外, 化合物12b对正常BEAS-2B细胞和MCF-10A细胞没有毒性。这些结果说明将二个活性亚单元拼接能够得到抗肿瘤活性更强的缀合物, 这为今后发展新型、高效的喹诺酮类抗肿瘤药物提供了新的思路。
实验部分BrukerAM-400Hz型核磁共振仪(TMS内标, CDCl3为溶剂); MINQI∧OSLl002N电子天平(上海民桥精密科学仪器有限公司); DF-101S集热式恒温加热磁力搅拌器(河南巩义市予华仪器有限责任公司); XR4显微熔点测定仪(上海光学仪器厂); 超净工作台(北京半导体设备一厂); BB16/BB5060仪器CO2培养箱(上海力创科学仪器有限公司); ELx800通用酶标仪(美国BioTek公司); CKX31型倒置显微镜(奥林巴斯公司)。
环丙沙星、SAHA、相应的二酸、取代苯胺、BBr3、三氟乙酸、EDC、DMAP均购自百灵威科技有限公司(质量分数≥98%); 所用的其他试剂购自国药集团化学试剂有限公司(质量分数≥95%); DMEM、MEM、RPMI-1640培养基、胎牛血清购自Hyclone公司; 人大细胞肺癌NCI-H460、乳腺癌MCF-7、肺癌A549、人口腔表皮样癌KB和宫颈癌HeLa细胞株以及人正常肺上皮细胞BEAS-2B和人正常乳腺上皮细胞MCF-10A购自武汉大学细胞典藏中心, 本实验室冻存使用。
1 化学合成 1.1 庚(辛、壬)二酸酐(2a~2c)的合成称取相应的二酸化合物1 (19.3 mmol)于25 mL单口瓶中, 加入8 mL乙酸酐, 加热回流1 h后, 减压脱溶剂得到相应的酸酐化合物2。
庚二酸酐(2a):白色固体, 产率: 96.6%, mp 51~53 ℃; 1H NMR (400 MHz, CDCl3): δ2.41 (m, 2H), 2.34 (t, J=7.6 Hz, 2H), 1.71 (t, J=7.2 Hz, 2H), 1.63 (m, 2H), 1.42 (m, 2H)。
辛二酸酐(2b):白色固体, 产率: 95.4%, mp 52~55 ℃; 1H NMR (400 MHz, CDCl3): δ2.47 (t, J=7.6 Hz, 4H), 1.56 (t, J=7.2 Hz, 4H), 1.34 (m, 4H)。
壬二酸酐(2c):白色固体, 产率: 97.7%, mp 59~61 ℃; 1H NMR (400 MHz, CDCl3): δ2.42 (t, J=7.6 Hz, 2H), 2.34 (m, 2H), 1.69 (t, J=6.8 Hz, 3H), 1.61 (t, J=6.4 Hz, 3H), 1.35 (m, 4H)。
1.2 苯胺甲酰基己(庚、辛)酸化合物(5、6)的合成氩气保护下, 将庚(辛、壬)二酸酐2 (9.8 mmol)溶于无水四氢呋喃中(35 mL), 加入N-Boc-对苯二胺(2.041 g, 9.8 mmol)或4-甲氧基苯胺(1.207 g, 9.8 mmol)后, 在室温下继续反应3 h。减压脱溶剂得到粗产物, 经硅胶柱纯化(二氯甲烷-甲醇60:1, v/v)得到了白色的固体5、6。
6-(4-N-Boc-氨基)苯胺甲酰基己酸(5a):白色固体, 产率: 91.2%, mp 105~107 ℃; 1H NMR (400 MHz, acetone-d6): δ12.19 (s, -COOH, 1H), 9.61 (s, -NH, 1H), 9.35 (s, -NH, 1H), 7.46 (d, J=8.8 Hz, 2H), 7.07 (d, J=8.0 Hz, 2H), 2.40 (m, 2H), 2.31 (t, J=6.8 Hz, 2H), 1.71 (t, J=6.8 Hz, 2H), 1.62 (t, J=7.2 Hz, 2H), 1.57 (s, 9H), 1.41 (m, 2H)。
7-(4-N-Boc-氨基)苯胺甲酰基庚酸(5b):白色固体, 产率: 89.6%, mp 114~116 ℃; 1H NMR (400 MHz, acetone-d6): δ12.25 (s, -COOH, 1H), 9.63 (s, -NH, 1H), 9.58 (s, -NH, 1H), 7.47 (d, J=8.0 Hz, 2H), 7.04 (d, J=8.4 Hz, 2H), 2.32 (m, 4H), 1.58 (s, 9H), 1.57 (m, 4H), 1.33 (m, 4H)。
8-(4-N-Boc-氨基)苯胺甲酰基辛酸(5c):白色固体, 产率: 93.4%, mp 118~111 ℃; 1H NMR (400 MHz, acetone-d6): δ12.23 (s, -COOH, 1H), 9.61 (s, -NH, 1H), 9.33 (s, -NH, 1H), 7.49 (d, J=8.4 Hz, 2H), 7.02 (d, J=8.4 Hz, 2H), 2.39 (m, 2H), 2.31 (t, J=7.6 Hz, 2H), 1.68 (m, 3H), 1.59 (t, J=6.4 Hz, 3H), 1.57 (s, 9H), 1.33 (m, 4H)。
6-(4-甲氧基)苯胺甲酰基己酸(6a):白色固体, 产率: 88.9%, mp 77~79 ℃; 1H NMR (400 MHz, acetone-d6): δ12.04 (s, -COOH, 1H), 9.36 (s, -NH, 1H), 7.45 (d, J=8.8 Hz, 2H), 7.01 (d, J=8.8 Hz, 2H), 3.81 (s, 3H), 2.27 (m, 2H), 2.18 (m, 2H), 1.59 (t, J=7.6 Hz, 2H), 1.47 (t, J=7.2 Hz, 2H), 1.31 (m, 2H)。
6-(4-甲氧基)苯胺甲酰基庚酸(6b):白色固体, 产率: 86.5%, mp 82~84 ℃; 1H NMR (400 MHz, acetone-d6): δ11.98 (s, -COOH, 1H), 9.33 (s, -NH, 1H), 7.38 (d, J=8.4 Hz, 2H), 6.69 (d, J=9.2 Hz, 2H), 3.78 (s, 3H), 2.35 (t, J=7.6 Hz, 2H), 2.29 (m, 2H), 1.67 (t, J=7.2 Hz, 2H), 1.59 (t, J=7.6 Hz, 2H), 1.39 (m, 4H)。
6-(4-甲氧基)苯胺甲酰基庚酸(6c):白色固体, 产率: 92.7%, mp 91~93 ℃; 1H NMR (400 MHz, acetone-d6): δ12.01 (s, -COOH, 1H), 9.32 (s, -NH, 1H), 7.40 (d, J=8.0 Hz, 2H), 7.03 (d, J=8.8 Hz, 2H), 3.79 (s, 3H), 2.41 (t, J=7.6 Hz, 2H), 2.29 (t, J=7.2 Hz, 2H), 1.67 (m, 3H), 1.58 (t, J=7.2 Hz, 3H), 1.36 (m, 4H)。
1.3 4-羟基苯胺甲酰基己(庚、辛)酸化合物(7)的合成氩气保护下, 将化合物6 (7.9 mmol)溶解到CH2Cl2 (25 mL)中, -20 ℃下滴加BBr3 (2.2 mL, 23.7 mmol), 继续反应12 h, 加入2 mL甲醇淬灭反应, 用乙酸乙酯(3 × 40 mL)萃取, 合并有机层后加入50 mL饱和NaHCO3溶液洗涤, 无水硫酸钠干燥, 减压脱溶剂得到粗产物, 经硅胶柱纯化(石油醚-乙酸乙酯1:1, v/v)得到化合物7。
6-(4-羟基)苯胺甲酰基己酸(7a):白色固体, 产率: 84.6%, mp 82~85 ℃; 1H NMR (400 MHz, acetone-d6): δ12.15 (s, -COOH, 1H), 9.32 (s, -NH, 1H), 9.11 (s, -OH, 1H), 7.41 (d, J=8.0 Hz, 2H), 6.99 (d, J=8.4 Hz, 2H), 2.26 (t, J=7.2 Hz, 2H), 2.17 (t, J=7.6 Hz, 2H), 1.60 (t, J=7.2 Hz, 2H), 1.46 (t, J=7.6 Hz, 2H), 1.33 (m, 2H)。
6-(4-羟基)苯胺甲酰基庚酸(7b):白色固体, 产率: 86.5%, mp 87~90 ℃; 1H NMR (400 MHz, acetone-d6): δ9.35 (s, -NH, 1H), 9.14 (s, -OH, 1H), 7.43 (d, J=8.0 Hz, 2H), 7.02 (d, J=8.4 Hz, 2H), 2.38 (m, 2H), 2.28 (t, J=7.6 Hz, 2H), 1.66 (t, J=7.6 Hz, 2H), 1.58 (t, J=6.4 Hz, 2H), 1.36 (m, 4H)。
6-(4-羟基)苯胺甲酰基庚酸(7c):白色固体, 产率: 87.8%, mp 94~96 ℃; 1H NMR (400 MHz, acetone-d6): δ12.12 (s, -COOH, 1H), 9.33 (s, -NH, 1H), 9.08 (s, -OH, 1H), 7.46 (d, J=8.8 Hz, 2H), 6.98 (d, J=8.4 Hz, 2H), 2.43 (m, 2H), 2.30 (t, J=7.6 Hz, 2H), 1.68 (t, J=6.8 Hz, 3H), 1.59 (m, 3H), 1.34 (m, 4H)。
1.4 4-氨基苯胺甲酰基己(庚、辛)酸化合物(8)的合成将化合物5 (8.2 mmol)溶于CH2Cl2中(15 mL), 0 ℃下加入5 mL TFA, 继续反应1 h后, 用饱和NaHCO3 (20 mL)洗涤, 无水硫酸钠干燥, 减压脱溶剂得到粗产物, 经硅胶柱纯化(二氯甲烷-甲醇60:1, v/v)得到化合物8。
6-(4-氨基)苯胺甲酰基己酸(8a):白色固体, 产率: 89.5%, mp 72~74 ℃; 1H NMR (400 MHz, acetone-d6): δ12.23 (s, -COOH, 1H), 9.35 (s, -NH, 1H), 7.51 (d, J=8.4 Hz, 2H), 7.08 (d, J=8.0 Hz, 2H), 5.09 (s, 2H, -NH2), 2.39 (t, J=7.2 Hz, 2H), 2.33 (t, J=7.2 Hz, 2H), 1.69 (m, 2H), 1.58 (t, J=7.6 Hz, 2H), 1.39 (m, 2H)。
7-(4-氨基)苯胺甲酰基庚酸(8b):白色固体, 产率: 90.7%, mp 76~78 ℃; 1H NMR (400 MHz, acetone-d6): δ12.25 (s, -COOH, 1H), 9.33 (s, -NH, 1H), 7.49 (d, J=8.4 Hz, 2H), 7.06 (d, J=8.0 Hz, 2H), 5.11 (s, 2H, -NH2), 2.34 (m, 2H), 2.31 (t, J=7.2 Hz, 2H), 1.60 (t, J=7.2 Hz, 2H), 1.56 (t, J=6.8 Hz, 2H), 1.35 (m, 4H)。
8-(4-氨基)苯胺甲酰基辛酸(8c):白色固体, 产率: 93.4%, mp 84~86 ℃; 1H NMR (400 MHz, acetone-d6): δ12.19 (s, -COOH, 1H), 9.31 (s, -NH, 1H), 7.52 (d, J=8.4 Hz, 2H), 7.05 (d, J=8.4 Hz, 2H), 5.13 (s, 2H, -NH2), 2.41 (t, J=7.6 Hz, 2H), 2.29 (t, J=7.2 Hz, 2H), 1.69 (m, 3H), 1.61 (t, J=6.8 Hz, 3H), 1.37 (m, 4H)。
1.5 羧酸类环丙沙星-HDACi缀合物(10、11)的合成氩气保护下, 将化合物7或8 (3.1 mmol)和环丙沙星(1.027 1 g, 3.1 mmol)溶解到CH2Cl2 (20 mL)中, 0 ℃下加入4-二甲氨基吡啶(DMAP, 37 mg, 0.3 mmol)或者1-羟基苯并三唑(HOBT, 419 mg, 3.1 mmol), 反应5 min后, 加入1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐(EDC×HCl, 1.114 1 g, 6.2 mmol), 撤掉冰浴在室温反应24 h后, 向反应液中加入20 mL蒸馏水, 用二氯甲烷(3 × 20 mL)萃取, 有机层用无水Na2SO4干燥, 脱除溶剂, 经硅胶柱纯化(二氯甲烷-甲醇15:1~9:1, v/v)得到了相应的缀合物10、11。
1.6 肟酸类环丙沙星-HDACi缀合物(12、13)的合成氩气保护下, 0 ℃下将羧酸化合物10、11(0.5 mmol)溶解到THF (10 mL)中, 搅拌10 min后, 向反应液中加入10 mL新制备的羟胺溶液, 缓慢升至室温后继续反应1 h, 减压除去溶剂, 经硅胶柱纯化(二氯甲烷-甲醇30:1~10:1)得到了异肟酸化合物12、13。羟胺溶液的制备:将盐酸羟胺(333.6 mg, 4.8 mmol)和氢氧化钾(268.8 mg, 4.8 mmol)加入到甲醇(15 mL)中, 升温至40 ℃反应15 min, 析出大量的沉淀, 将反应液冷却至室温, 滤除沉淀, 所得滤液即是新制备的羟胺溶液。
1.7 2-苯酰胺类环丙沙星-HDACi缀合物(14、15)的合成氩气保护下, 将羧酸缀合物10、11(2.9 mmol)、苯并三氮唑-1-基氧基三(二甲基氨基)磷鎓六氟磷酸盐(BOP试剂, 1.954 9 g, 4.4 mmol)和三乙胺(1.335 7 mg, 13.2 mmol)溶解到无水DMF (50 mL)中, 室温反应30 min后, 加入邻苯二胺(314 mg, 2.9 mmol), 继续反应12 h, 向反应液中加入50 mL蒸馏水, 用乙酸乙酯(3 × 20 mL)萃取, 有机层用无水Na2SO4干燥, 脱除溶剂, 经硅胶柱纯化(二氯甲烷-甲醇30:1~10:1, v/v)得到2-苯酰胺类缀合物14、15。
2 HDACs抑制活性实验使用HDAC试剂盒测试环丙沙星-HDACi缀合物对HDAC1、HDAC2、HDAC6的抑制活性, 以SAHA为阳性对照药。按照试剂盒操作说明书在测试板中每孔加入BSA、HDAC荧光底物、HDAC酶(HDAC1、HDAC2、HDAC6)和不同浓度的待测物。将测试板在37 ℃下反应30 min后, 每孔再加入HDAC Developer, 并继续在37 ℃下放置15 min, 使用酶标仪在359 nm和440 nm波长测定每孔的荧光值, 计算IC50值。
3 分子对接HDAC1晶体结构(PDB code: 4BKX)由Protein Data Bank下载, HDAC6的晶体结构目前还未解析出来, 参照文献以HDAC8晶体结构(PDB code: 1T69)为模板对HDAC6进行同源建模[19-21]。通过ChemBio 3D ultra 14.0和Autodock4.2D中的Ligand模块进行小分子结构预处理。使用Autodock4.2D对接软件进行对接[22], 口袋盒子以晶体结构中的配体定义, 其对接盒子边长设置为30 Å, 并使用半经验自由能进行评价, 拉马克遗传算法循环为80次, 其他参数保持默认。
4 抗增殖活性实验选取人大细胞肺癌NCI-H460、乳腺癌MCF-7、肺癌A549、人口腔上皮样癌KB和人宫颈癌HeLa细胞为测试细胞株。以SAHA为阳性对照药, 采用CCK-8试剂盒对合成的化合物进行抗肿瘤活性评价。取对数生长期的测试细胞株悬浮于含10%胎牛血清的培养基中, 铺至96孔培养板中。待细胞完全贴壁后, 弃去原培养液, 加入10 µL的含有测试药物的培养液培养24 h, 弃去原培养液, 每孔加入10 μL CCK-8溶液, 在培养箱中继续孵育4 h后, 用酶标仪在490 nm波长测定每孔的吸光度值, 分析实验结果, 并计算出IC50。
| [1] | Luo J, Su F, Chen D, et al. Deacetylation of p53 modulates its effect on cell growth and apoptosis[J]. Nature, 2000, 408 :377–381. DOI:10.1038/35042612 |
| [2] | Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer[J]. Nat Rev Cancer, 2006, 6 :38–51. DOI:10.1038/nrc1779 |
| [3] | Tang YM, Huang WY, Yu NF. Structure-activity relationships of histone deacetylase inhibitors[J]. Acta Pharm Sin (药学学报), 2009, 44 :1072–1083. |
| [4] | Yao YW, Yao HQ, Jiang S, et al. Progress in clinical study of histone deacetylases inhibitors as anticancer agents[J]. Chin New Drugs J (中国新药杂志), 2013, 22 :294–299. |
| [5] | Lai MJ, Huang HL, Pan SL, et al. Synthesis and biological evaluation of 1-arylsulfonyl-5-(N-hydroxyacrylamide) indoles as potent histone deacetylase inhibitors with antitumor activity in vivo[J]. J Med Chem, 2012, 55 :3777–3791. DOI:10.1021/jm300197a |
| [6] | Wagner FF, Olson DE, Gale JP, et al. Potent and selective inhibition of histone deacetylase 6(HDAC6) does not require a surface-binding motif[J]. J Med Chem, 2013, 56 :1772–1776. DOI:10.1021/jm301355j |
| [7] | Lee HY, Tsai AC, Chen MC, et al. Azaindolylsulfonamides, with a more selective inhibitory effect on histone deacetylase 6 activity, exhibit antitumor activity in colorectal cancer HCT116 cells[J]. J Med Chem, 2014, 57 :4009–4022. DOI:10.1021/jm401899x |
| [8] | Zhang ZP, Li DH, Gu QQ, et al. The research progress of selective histone deacetylase inhibitors[J]. Chin J Med Chem (中国药物化学杂志), 2013, 23 :321–330. |
| [9] | Lin KJ, Liu ZH, You QD. QSAR studies of antitumor inhibitors of histone deacetylases[J]. J China Pharm Univ (中国药科大学学报), 2004, 35 :106–109. |
| [10] | Marks PA. Discovery and development of SAHA as an anticancer agent[J]. Oncogene, 2007, 26 :1351–1356. DOI:10.1038/sj.onc.1210204 |
| [11] | Gryder BE, Rood MK, Johnson KA, et al. Histone deacetylase inhibitors equipped with estrogen receptor modulation activity[J]. J Med Chem, 2013, 56 :5782–5796. DOI:10.1021/jm400467w |
| [12] | Katsarou ME, Efthimiadou EK, Psomas G, et al. Novel copper (Ⅱ) complex of N-propyl-norfloxacin and 1, 10-phenanthroline with enhanced antileukemic and DNA nuclease activities[J]. J Med Chem, 2008, 51 :470–478. DOI:10.1021/jm7013259 |
| [13] | Hu GQ, Wu XK, Wang X, et al. Synthesis and antitumor activity of C3 heterocyclic-substituted fluoroquinolone derivatives (I):ciprofloxacin aminothiodiazole Schiff-base[J]. Acta Pharm Sin (药学学报), 2008, 43 :1112–1115. |
| [14] | Hu GQ, Huo LL, Wang GQ, et al. Part IV:design, synthesis and antitumor activity of fluoroquinolone C-3 heterocycles:bis-oxadiazole methylsulfide derivatives derived from ciprofloxacin[J]. Acta Pharm Sin (药学学报), 2012, 47 :1017–1022. |
| [15] | Li J, Jin Y, Shou KJ, et al. Design, synthesis and antitumor activity of quinolone derivatives[J]. Chin Pharm J (中国药学杂志), 2013, 48 :2055–2060. |
| [16] | Azéma J, Guidetti B, Dewelle J, et al. 7-((4-Substituted) piperazin-1-yl) derivatives of ciprofloxacin:synthesis and in vitro biological evaluation as potential antitumor agents[J]. Bioorg Med Chem, 2009, 17 :5396–5407. DOI:10.1016/j.bmc.2009.06.053 |
| [17] | Xu QJ, Huo LL, Wu XK, et al. Synthesis and antitumor activity of ciprofloquinolone bis-(C3/C7 hydrazone) s[J]. J China Pharm Univ (中国药科大学学报), 2013, 44 :35–38. |
| [18] | Itoh Y, Suzuki T, Miyata N. Isoform-selective histonedeacetylase inhibitors[J]. Curr Pharm Des, 2008, 14 :529–544. DOI:10.2174/138161208783885335 |
| [19] | Senger J, Melesina J, Marek M, et al. Synthesis and biological investigation of oxazole hydroxamatesas highly selective histone deacetylase 6(HDAC6) inhibitors[J]. J Med Chem, 2016, 59 :1545–1555. DOI:10.1021/acs.jmedchem.5b01493 |
| [20] | UniProt C. Activities at the universal protein resource (UniProt)[J]. Nucleic Acids Res, 2014, 42 :D191–D198. DOI:10.1093/nar/gkt1140 |
| [21] | Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega[J]. Mol Syst Biol, 2011, 7 :539. |