 2016, Vol. 51
2016, Vol. 51

替格瑞洛是抑制血小板聚集的药物,由阿斯利康公司研制,于2010和2011年分别在欧盟和美国上市。它的作用靶标是二磷酸腺苷 (ADP) 受体P2Y12,与已经上市的血小板抑制剂氯吡格雷等作用靶标相同,但结合位点不同,氯吡格雷在P2Y12受体与ADP竞争结合位点,发生不可逆结合; 而替格瑞洛可逆性地结合于P2Y12变构区。
2 研制背景与靶标血小板的生理功能是止血,血管破裂出血,经血小板聚集促使血凝。然而血小板在动脉壁斑块上的聚集会形成血栓,造成栓塞,可发生危及生命的心肌梗死和脑卒中。所以防止血栓形成是降低心脑血管疾病的重要环节。
血小板上具有核苷受体,其中P2Y12受体亚型,经ADP结合而活化,在动脉血栓形成的过程中起关键作用。ADP结合于血小板的P2Y12受体,使血小板形状改变,暴露出糖蛋白IIb/IIIa (IIb/IIIa为血小板与纤维蛋白原发生交联的位点)。再经内源性聚集调节剂如血栓烷A2、5-HT和ADP等的释放,发生放大效应,形成持续性的血小板聚集。
3 评价活性的模型化合物抑制血小板聚集的初筛模型是用离体实验。将人血小板经洗涤后悬浮于Tyrode缓冲液中,加入ADP以使血小板聚集,加入不同浓度的受试物以抑制聚集过程,用浊度仪测定聚集程度,计算受试物的活性IC50或pIC50 (Humphries RG,Tomlinson W,Ingall AH,et al. FPL 66096: a novel,highly potent and selective antagonist at human platelet P2T-purinoceptors. Br J Pharmacol,1994,113: 1057−1063)。
4 化学线索—ATP对P2Y12具有弱抑制作用ADP (1) 是P2Y12受体激动剂,而ATP (2) 对P2Y12具有弱抑制作用,pIC50 = 3.6,虽然ATP水溶性良好 (logD7.4 = −3),适于注射应用,但由于对其他受体亚型也有抑制作用,而且在体内的稳定性差,不能药用。但ATP可作为研发P2Y12拮抗剂的起始物。
5 结构改造对ATP的结构改造主要集中在3个部位,即三磷酸链、C2的取代和C6上的胺基 (N6) 取代,如通式3所示。
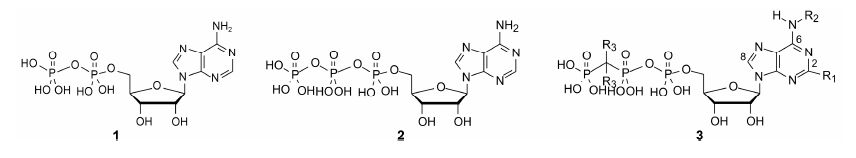
ATP比ADP多出一个γ-磷酸基,对P2Y12作用正好相反,由ADP的激动转变为ATP的拮抗作用,推测归因于ATP的γ-磷酸基负电荷起重要作用 (这是一个科学假定)。推论保持3个磷酸单元是维持拮抗作用之必需。然而在心血管组织细胞表面存在外核苷酸酶 (ectonucleotidase),可将ATP的γ-磷酸基水解生成ADP。为了阻止水解作用应增加第3个磷酸基的稳定性,因而须对ATP的结构修饰。
将连接β,γ-二磷酸之间的氧原子改换成-CH2-,提高了γ磷酸基对水解的抗性,化合物稳定性增高。然而,β,γ-CH2-的变换,使得未端质子的pKa提高到8.1 (酸性减弱),而原来的三磷酸基未端质子的pKa为6.6,以至于在生理条件下 (pH 7.4),β,γ-CH2-三磷酸未端的-OH不能离解成负离子,导致荷电状态类似于ADP的3个负电荷 (ATP有4个负电荷),因而降低了抑制活性。
当β,γ-CH2-的两个氢原子被卤原子置换 (通式3中R3=F或Cl),由于卤素的强电负性,使未端质子的pKa接近于ATP,活性也随之提高,例如化合物4的pIC50=3.5,与ATP相近。
5.2 C2的取代基于合成反应的考虑,将C8和C2 (通式3) 的H可用基团取代。结果表明,C8即使用最小的基团取代都会降低拮抗作用,因而C8-H不可变换。而C2-H被其他基团置换,可提高活性。例如2-甲硫基取代的ADP促进血小板聚集的活性强于ADP约30倍(Gough G,Maguire MH,Penglis F. Analogues of adenosine 5¢-diphosphate. New platelet aggregators. Influence of purinering and phosphate chain substitutions on the platelet taggregating potency of adenosine 5¢- diphosphate. Aust Mol Pharmacol,1972,8: 170−177)。C2为乙硫基或正丙硫基取代的化合物,抑制血小板聚集的活性比4强10 000倍,例如化合物5的pIC50 = 8.6,6的pIC50 = 8.16。提示在C2处引入疏水基团提高抑制活性。然而再延长C2的疏水链,不能提高活性。
因化合物5对P2Y12的强抑制活性和选择性,确定为进入开发的候选化合物。用麻醉大鼠、犬和人进行体内实验,化合物5半衰期很短,t1/2 = 2 min。静脉灌注0.1 mg·kg−1·min−1,15 min后完全抑制了血小板聚集,停药后15 min则完全恢复。化合物5的这种快速起效和快速消失的特点,对于处置急性栓塞的患者是有利的。
5.3 N6的取代N6被双烷基取代,抑制活性锐减,故不宜双取代。而单烷基取代比未取代的化合物活 性高,链中可有杂原子取代,但以3~4个疏水性原子为最佳,烷基链延长会导致长时间作用而不利。经优化发现N6被CH2CH2SCH3单取代为佳,C2连接SCH2CH2CF3的化合物7,pIC50 = 9.35,比化合物5的活性强6倍,给药起效后20 min,复原率为79%,作用时间适宜。遂作为注射用药进入临床研究,命名为砍格雷洛 (7,cangrelor),为超短时抗血栓生成药物,目前处于Ⅲ期阶段 (Ingall AH,Dixon J,Bailey A,et al. Antagonists of the platelet P2T receptor: a novel approach to antithrombotic therapy. J Med Chem,1999,42: 213−220)。
6 P2Y12口服抑制剂 6.1 嘌呤苷酸的再改造在由起始物ATP设计P2Y12抑制剂中还实施了另一条路径,即将三磷酸 侧链用二羧酸置换,模拟三磷酸端基的负电荷,制备了化合物8,8虽然有活性,但低于7上百倍。进而对嘌呤环作结构变换,将C8换成N原子得到的化合物9抑制P2Y12的活性提升到与7相当,表明三唑并嘧啶替换嘌呤母核,活性提高百倍。进而用环戊基替换核糖,化合物10仍然保持了活性。由此确定了氮杂嘌呤 (即三唑并嘧啶) 与环戊基形成的类核苷成为P2Y12抑制剂新的骨架结构。
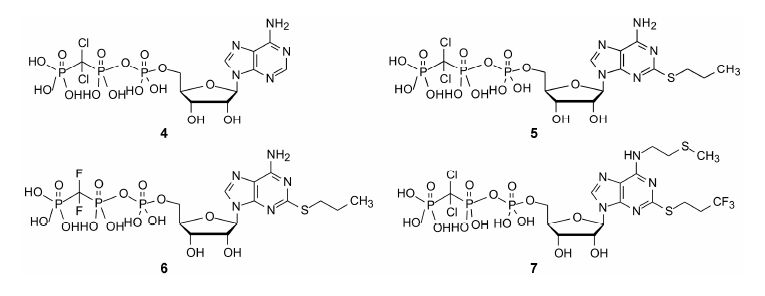
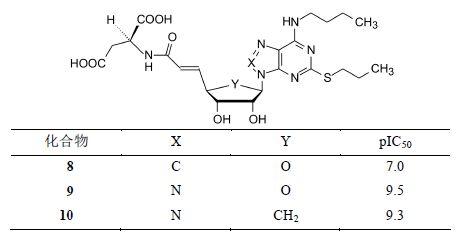
化合物9和10的活性虽然较高,但相对分子质量大于500,灌胃大鼠迅速从胆汁中排出,这不符合口服的要求,而且由于含有可形成两个负电荷的羧基和两个羟基,这些都是不利于口服吸收的结构因素,因而需“削减”该酸性侧链。
6.2 用受体结合实验评价高活性化合物前述的活性是评价化合物抑制ADP诱导洗涤过的血小板的聚集作用,属于功能性实验。为了进一步评价化合物的活性,用放射性125I标记的P2Y12拮抗剂被受试化合物从洗涤过的血小板上置换下来的结合力实验计算受试物pKi值。用已有的化合物证明该放射性置换实验值pKi与功能性实验值pIC50是平行的。
6.3 简化酸性侧链成中性短链仍然有效用组合合成的方法制备含有各种酸性侧链的化合物,意外地发现单羧酸化合物11仍具有活性,这显然与所谓的“γ-磷酸假定”相悖,更有意义的是,即使没有羧基的化合物12和13,仍然有一定的抑制活性,这为减小分子尺寸和改善药代性质提供了优化线索。不过化合物11~13的大鼠生物利用度和在体内的存留时间仍然较低。
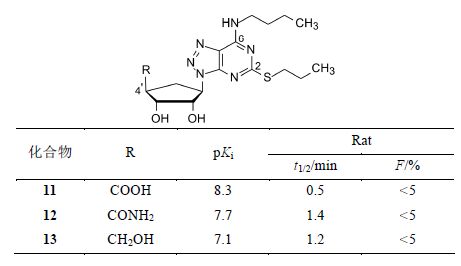
由于4' 位置的取代基发生了较大的变化,需要进一步优化C2和N6取代基,阿斯利康公司为此合成了6 000多个化合物,足见由注射剂变为口服用药的的艰巨性 (Bonnert RV,et al. PCT Int. Appl. W0199828300,1998; Chem Abstr.,1998,129: 95506; Guile SD,et al. PCT Int. Appl. WO199905143,1999; Chem Abstr.,1999,130: 168386)。化合物的结构与活性的关系归纳如下: ① 将4' 的R固定为COOH、CONH2或CH2OH,变换C2的连接基,此时疏水性侧链有利于提高抑制作用,其中正丙硫基的活性最强,若硫原子换成极性的氧或氮原子,则活性降低。因而将C2的连接基固定为正丙硫基。② N6连接疏水性基团也有利于提高活性,极性的侧链使活性降低; N连接两个烷基链成为叔胺对活性不利。反式2-苯基环丙基取代的化合物 活性明显强于其他侧链,例如化合物14~16,比相应的正丁基化合物11~13的活性强10倍。此外,化合物16的半衰期和口服生物利用度 (F = 35%) 也显著优于其他化合物。③ 化合物14~16含有两个手性中心,消旋体经拆分后得到的旋光异构体1R,2S的活性强于1S,2R对映体。
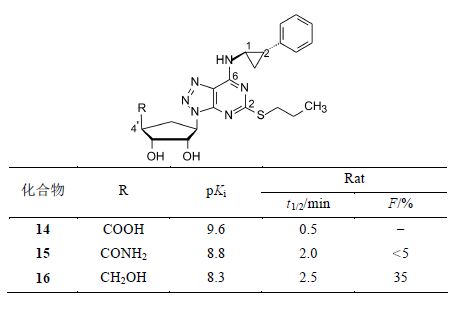
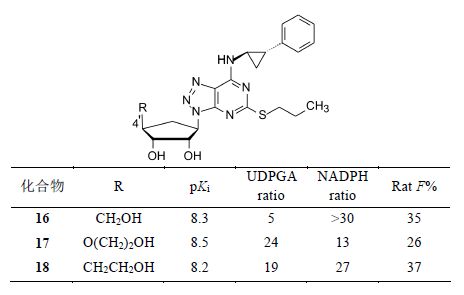
为了优化药代动力学性质,评价了化合物对Ⅰ相和Ⅱ相代谢的稳定性。建立的体外模型是在化合物与肝微粒体的温孵中加入NADPH (氧化代谢的辅酶) 和UDPGA (葡醛酸苷化的辅酶),同时加入可发生氧化代谢的药物齐留通 (zileuton) 和发生葡醛酸苷化的美沙芬 (dextromethorphan),当受试物可耐受大于20倍齐留通和10倍美沙芬的代谢的程度时,预示化合物在体内肝脏发生两相代谢都不高于10%,即小于2 mL·min−1·kg−1 (Springthorpe B,Bailey A,Barton P,et al. From ATP to AZD6140: the discovery of an orally active reversible P2Y12 receptor antagonist for the prevention of thrombosis. Bioorg Med Chem Lett,2007,17: 6013−6018)。
6.6 提高代谢稳定性—环戊基的4' 位取代基的优化化合物16对P2Y12的抑制活性以及大鼠的生物利用度和半衰期达到了一定的要求,然而用大鼠、犬和人肝细胞研究16的代谢行为时,发现大鼠对4'-CH2OH发生氧化代谢,而犬和人则是Ⅱ相的葡醛酸苷化。为了提高化合物对氧化和葡醛酸苷化的稳定性,对4'的R取代基进行优化,其中化合物17和18对Ⅱ相和Ⅱ相代谢达到稳定性的标准 (分别达到预设的比值),活性和生物利用度也基本达到要求。
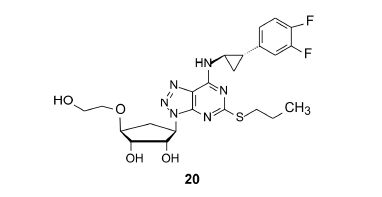
为了进一步优化体外活性和代谢稳定性,N6侧链上的苯环用不同的基团取代,化合物19~21呈现优良性质,表明苯环上有两个氟原子取代是有利的。化合物20的硫醚链端基CH3被CF3置换,对P2Y12的亲和力提高了0.5个对数单位,pKa达到9.2,活性最高。
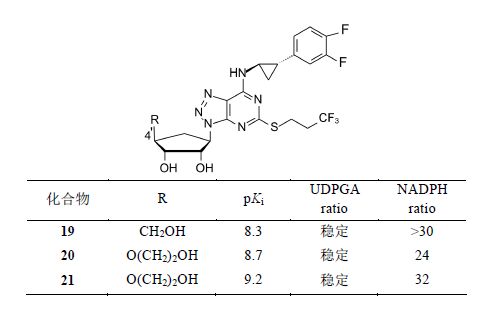
化合物19~21对大鼠和犬的药代动力学实验表明,20的口服生物利用度F大鼠 = 24%,F犬 = 72%,高于化合物19和21,综合药效学和药动学数据,预测化
合物20用于临床的治疗剂量最小,因而选定为候选药物,命名为替格瑞洛 (ticagrelor) 进入开发阶段。经Ⅲ期临床研究证明是急性冠状动脉综合征不稳定性心绞痛和心肌梗死的有效药物,在2010和2011年分别在欧盟和美国批准上市 (Springthorpe B,Bailey A,Barton P,et al. From ATP to AZD6140: the discovery of an orally active reversible P2Y12 receptor antagonist for the prevention of thrombosis. Bioorg Med Chem Lett,2007,17: 6013−6018)。
替格瑞洛虽然不是第一个上市的P2Y12受体拮抗剂,但作用于受体的变构区域,有别于它的先行者,结构类型也完全不同,也可认为是个首创性药物。阿斯利康公司研制成功替格瑞洛,合成了6 000多个目标化合物,其中的曲折复杂过程笔者不得而知,不过仅从研发过程中构建和精修化学结构,也可启示人们开拓思路。以下是研发中的重要节点: ① 发现ATP对P2Y12受体具有弱抑制作用,成为研究的起始点。② 在嘌呤环的C2和N6处引入疏水性侧链,提高了与受体的结合力。③ 修饰三磷酸基的βγ连接基,提高稳定性,并维持了必要的酸性。④ 骨架迁越用三唑并嘧啶替换嘌呤环,使活性提高逾百倍,此时分子的结合取向很可能发生了变化。该阶段研发出注射用的坎格雷拉 (现处于Ⅲ期临床)。⑤ 继续研究,发现了没有酸性侧链的拮抗剂。⑥ N6侧链上引入反式2-苯基环丙胺片段,活性提高一个数量级。⑦ 为提高代谢稳定性在环戊基的4' 位优化得到代谢稳定的基团。⑧ 精修苯环,引入二氟原子提高了活性和代谢稳定性,建成替格瑞洛分子。