 2016, Vol. 51
2016, Vol. 51



“十三五重大新药创制”倡导和鼓励研制首创性药物, 标志着国家发展到新的水平, 也体现了当今临床与市场对新药的需求。本世纪以来的跟进式新药明显减少, 彰显了首创药物的主导地位[1]。以靶标为核心的药物创新模式, 是以分子和细胞生物学基础研究为引擎, 发现新的生物功能分子, 过渡到以化学生物学为主导的探针与靶标分子的互动, 再引申到与药物化学对接, 进入新药研发的轨道。在这探针-先导物-候选物-新药批准的全过程中, 靶标的确证贯穿于始终。这是一条“定型”的主流研发模式, 本文不拟展开讨论, 而是从另一角度探索研发新药的思路。
在创新思维中有一种方法叫做反向思维 (亦称逆向思维), 是对已成定论的司空见惯的事物或观点反过来思考的一种思维方法, 即思维向相反面或对立面进行探索。人们在解决问题时, 往往习惯于按照熟悉的常规思维路径去思考, 这种从众的趋向就是正向思维, 形成思维定势。例如某靶标蛋白的高表达与某疾病的发生相关联, 多数人会在思维定势驱使下, 思路、策略乃至方法不约而同。其实, 实行反向思维有时也能得到意想不到的结果。本文试举几个实例讨论运用反向思维进行新药研究的模式。
1 阿片δ受体拮抗剂—依卢多林治疗腹泻型肠易激综合征提到阿片受体, 人们就会联想到镇痛药的研究。其实阿片受体 (包括δ、μ和κ等亚型, 这里不深入展开) 参与镇痛、抑制肠胃蠕动、呼吸抑制、心肌保护和免疫反应等多种生理活动。除呼吸抑制外, 引起这些生理表型的变化对于新药研究而言, 是正能量还是负能量, 取决于药物研发的目标。内源性配体例如脑啡肽或药物 (如吗啡) 可以作用于不同部位的受体, 对中枢神经的阿片受体是调节痛觉, 对外周的受体例如可抑制胃肠道蠕动。所以, 以镇痛为目标的阿片受体的选择性激动剂, 需要消除对消化道蠕动的抑制作用, 避免引起便秘的不良反应。但从反向思考, 若只抑制肠道的阿片受体, 并且避免进入中枢, 可成为没有镇痛和成瘾性的治疗胃肠道功能紊乱的药物。
强生公司设定的研究目标是口服的阿片受体拮抗剂, 作用靶标是肠壁的阿片受体, 最好不吸收进入血液循环, 更不要穿越血脑屏障进入中枢的非肽化合物。他们以内源性配体脑啡肽 (1, enkephalin) 为起始物, 经结构优化和构效关系研究, 最后研制成功了依卢多林 (2, iluxadoline), 成为首创的对阿片受体μ激动、δ拮抗的双重调节剂, 口服治疗腹泻型肠易激综合征[2], 于2015年5月FDA批准上市。
依卢多林是阿片δ受体小分子拮抗剂, 完全消除了镇痛作用和成瘾性, 相对分子质量为556, 比通常的阿片受体激动剂分子尺寸大, 但研发者不介意分子量加大, 因为药物不需要穿越血脑屏障 (大尺寸分子不易穿越), 指望穿肠而过的局部作用或更为有利。此外还在分子中加入了游离的羧基和氨基, 从而在体内形成两性离子不易被细胞吸收 (这对于口服吸收的药物是非常禁忌的, 因为极性过强难以穿膜吸收), 这个性质满足了作用于肠壁膜受体的要求, 局部作用而起效, 生物利用度低, 可将不良反应降低到最低限度。依卢多林成为反向思维指导下的成功的范例[3]。
热休克蛋白90 (HSP90) 是在对细胞产生损伤 的多种外在刺激下, 细胞产生的一种应激蛋白, 对机体发挥很多重要的作用, 最主要的是以“分子伴侣”的身份出现, 对其他蛋白质和多肽起稳定化作用。HSP90还能使发生构象错误的蛋白质恢复正常, 并降解严重损伤的蛋白质等。
肿瘤细胞中存在高表达量的HSP90, 可能是由于肿瘤细胞的不断增殖需要它作为分子伴侣, 通过对效应蛋白构象的稳定化, 维护和稳定肿瘤的增殖过程, 因而研发HSP90抑制剂成为创制抗肿瘤药物的活跃领域。
另一方面, 脑中风患者的缺血区ATP匮乏是脑损伤的始动因素, 而神经元内蛋白质翻译停滞是脑缺血引起迟发性神经元死亡的标志。研究表明, 高表达的热休克蛋白可以对抗和消解缺血区的两大变化: 蛋白质翻译停滞和ATP供给匮乏, 抑制细胞凋亡和坏死, 其机制与抑制异常蛋白聚集、减少细胞毒性相关。因此, 诱导热休克蛋白表达有益于脑缺血的保护作用。这样, 对HSP90蛋白的抑制或激活, 分别成为抗肿瘤药物和神经保护药物研究的两个相反的方向。
基于此, 刘磊等[4]从分子生物学入手, 用Affymetrix基因芯片比对正常果蝇与致凋亡基因的果蝇表达谱, 再将差异基因输入到基因表达谱数据库 (connectivity map) 中, 与小分子药物引起的基因变化进行匹配, 找到25个药物, 用这些药物分别饲养果蝇, 其中α-阻断剂特拉唑嗪 (3, terazosine) 提高了果蝇的存活率, 是由于抑制凋亡的缘故。进而研究发现特拉唑嗪也抑制哺乳动物细胞凋亡。
为了解析特拉唑嗪的作用环节和机制, 合成的探针分子是它的类似物4, 4是将特拉唑嗪的喹唑啉环的N1用CH置换, 变为喹啉环母核, 分子的其他结构不变。实验表明4没有α-阻断作用, 但仍保持抗凋亡活性, 说明3不是经阻断α受体的机制起作用的。
用生化方法证明特拉唑嗪结合并激活了磷酸甘油酸激酶Pgk1, 促进ATP的释放, ATP提高了HSP90的伴侣蛋白 (ATP酶) 活性, 导致HSP90促进了对应激因子的抵抗。特拉唑嗪对果蝇、小鼠和大鼠的氧化应激和脑缺血都有保护作用。这个实例说明了基于对热休克蛋白研究的反向思维, 从基因谱比对到动物实验的概念验证, 涵盖了蛋白表达、酶动力学、细胞生物学、探针合成、晶体结构解析 (图 1) 等内容, 作了大胆的假设和科学的求证。由于特拉唑嗪已是临床常用的药物, 或许直接成为治疗或缓解脑卒中引起的损伤的药物。当然, 也可以作为高起点的先导物, 保持活性的同时, 优化结构以利于进入中枢神经系统的药代动力学性质。
 | 图 1 特拉唑嗪与HSP90结合的模式 |
心肌细胞膜上由hERG基因表达的钾通道在心脏动作电位的去极化过程起关键作用, 这在很大程度上是由于该通道有独特的门控特征, 即较慢的活化和迅速的电压控制去活化过程。化合物抑制hERG 钾通道可引起心律失常, 心电图呈现Q-T波延长, 表明心室从开始的去极化到最后的复极化之间延长了时间, 严重时可引起猝死。
新药研究中由于较常遇到抑制该通道的化合物, 而且也不限于作用于心脏药物 (例如抗过敏药物特非那定和西沙比利等, 因严重的心脏事件而停止使用), 因而在新药的早期研究阶段考察化合物对hERG通道是否有抑制作用已成必须实验的项目。
统计学表明对抑制hERG通道的化合物大大多于激动剂, 这是因为抑制剂大都结合于通道的内腔中, 而且亲和力强, 阻止了钾离子的传输。而激活剂则结合于通道的门控处, 是个取决于电压的迅速去活化过程, 调节钾的内流, 对分子结构有特异的要求[5]。
人们由于惯性思维, 往往期盼化合物远离对hERG通道作用, 避免对该通道的作用, 以至于较少从激活hERG通道入手, 研究治疗心律失常的药物, 例如治疗Q-T延长综合征等。不过一些公司已经注意到hERG激活剂的研究, 如安万特公司研究了化合物RPR-260243 (5)[6], 通过减低去活化和显著减缓通道关闭速率而加强了电流。用丙氨酸扫描突变技术证明化合物5结合的位点是胞浆末端的S5域的Leu553和Phe557以及S6域的Asp658和Val659残基。分子模拟提示其结合模式与抑制剂显著不同。此外, 辉瑞公司研究的化合物PD-118057 (6)[7]和雅培公司研究的ICA-105574 (7)[8]等都是hERG通道的激活剂。虽然迄今未见有临床研究的报道, 但循此反向路径不失为创新领域的探索。
胃动素 (motilin) 是由十二指肠和空肠黏膜的内分泌细胞合成的内源性激素二十二肽, 通过作用于胃幽门窦和十二指肠道细胞的胃动素受体, 引起平滑肌收缩, 并且通过周期性释放胃动素调节胃肠移行性复合波 (MMC)。
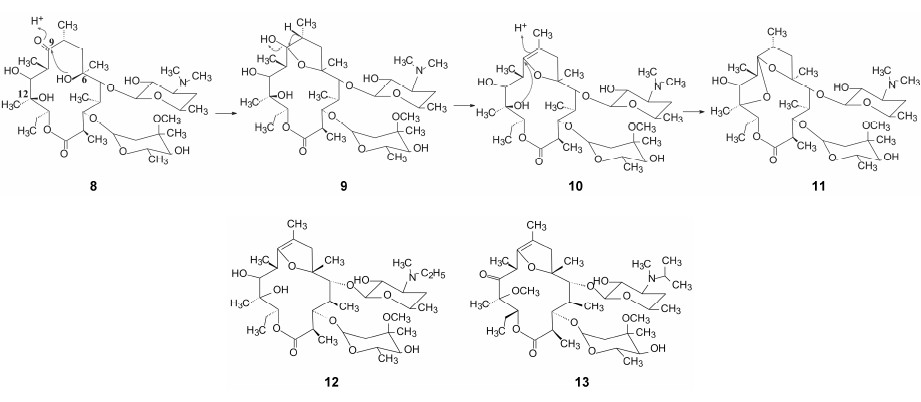
大环内酯类抗生素红霉素(8) 的不良反应是刺激胃肠道的蠕动、引起恶心与呕吐等, 因而后继的大环内酯消除了该不良反应。研究表明, 这种不良反应非常类似于胃动素的生理功能, 是由于与胃动素作用于同一受体。
研究表明, 12和16元大环内酯对胃动素受体的活性很弱, 只有14元环的红霉素具有这种胃肠道动力刺激作用。从反向考虑, 若利用红霉素这种类似于胃动素的药理作用, 有可能研制出新结构类型的胃动力药, 当然就需要消除它的抗菌活性。为实现该反向研究, 就需强化原有的副作用, 扬弃抗菌的主作用, 进行主副作用的升降转换。
红霉素 (8) 在酸性条件下, 6位羟基与9位酮基发生羟醛缩合, 生成环状半缩酮 (9), 后者与12位羟基失水 (10) 形成两个四氢呋喃环化合物 (11)。化合物9和11失去了抗菌作用, 提高了对胃肠道动力刺激作用。因而克拉霉素、阿奇霉素和泰利霉素的结构都避免了羟醛缩合的发生。
实施反向研究, Omura等合成了红霉素的改构 物, 伊屈西诺 (12, idremcinal)[9]、米特西诺 (13, mitemcinal)[10]都在大环上嵌入了二氢呋喃环, 显示胃动素受体的激动作用, 虽然这些候选药物迄今未进入III期临床阶段, 但不失为反向思维驱动创新药物的探索。
5 共价键药物药物与靶标发生共价键结合所呈现的药理作用, 由于结合键稳固, 大都为不可逆反应, 产生持久性作用。构成体内所有的成分都不含亲电性基团, 核酸、受体蛋白、酶、脂质乃至辅酶都含有亲核性基团或片段, 因而可发生共价结合的药物分子中都存在程度不同的亲电性基团。
最早的共价键药物多为抗癌或抗感染药物, 因为是以杀灭或清除外来的病原体为目标, 所以用烷化剂类抗肿瘤药物 (如氮芥和磺酸酯类)。这些药物有较强的亲电性而缺乏选择性, 广泛的脱靶作用成为敌我不分的“虎狼药”, 引起显著的毒副作用, 现大都已停用。也因此, 药物化学家长期以来回避共价键药物的研制, 特别是针对调节机体组织器官功能的所谓药效类药物 (pharmacodynamics, 为化疗药物chemotherapeutics之对) 是绝对要规避的。
随着有机物理化学、结构生物学和计算化学的发展, 发明了一批不限于用作化疗的共价键药物, 其来源或是幸运发现或是理性设计, 都意味着共价键药物的复兴, 现实状况不应再坚守拒绝含有亲电基团的惯性思维。作者以为把握两个环节是研制共价键药物的关键: 一是亲电基团在药物结构中所处的位置和连接的环境, 另一是电性的适度, 即药物的亲电性与靶标的亲核性的适配, 无过 (避免脱靶作用) 无不及 (稳固的共价结合)。换言之, 在适宜的位置 (形状契合、取向正确、距离合适) 发生“不温不火”的键合作用 (强度匹配)。以下列举有代表性的共价键药物, 简要解析其成功之所在。
5.1 阿司匹林百年前发明的阿司匹林, 自20世纪70年代发现前列腺素的功能后, 揭示其作用靶标是抑制环氧合酶 (COX), 阻止了下游的血栓烷A2的合成, 带来了防止血栓形成的新用途。阿司匹林是不可逆抑制剂, 结构中的乙酰基将COX的丝氨酸乙酰化, 发生共价结合。从有机化学视角分析, 在温和条件下高效率发生的这个酯交换反应, 似乎难以想象。其实, 由于乙酰水杨酸在酶活性中心的残基协同作用下, 提高了乙酰基的亲电性, 也提高了丝氨酸羟基的亲核性。图 2a是阿司匹林与COX活性部位的晶体结构投影图, 显示了苯环的π-π叠合, 羧基的静电引力, 乙酰基的氢键结合, 分子中似乎没有多余原子, 原子效率极高。图 2b是阿司匹林与COX结合的示意图和形成共价结合的过程。
 | 图 2 阿司匹林与COX活性部位的结合特征 (a) 及形成共价结合过程的示意图 (b) |
阿司匹林至今没有跟进的药物, 是不可替代的“me-only”药物。阿司匹林的发明者Hoffmann当初不会想到为缓解其父的关节炎疼痛而合成的阿司匹林是如此之完美, 以至于迄今无法超越 (no-better)。当今的科学发展人们理应进行理性设计优良的共价键药物。
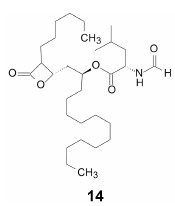
脂肪堆积导致肥胖。人体摄入的脂肪在肠道中被脂肪酶催化水解, 消化成游离脂肪酸或单酰基甘油, 吸收后再合成为脂肪。减肥药奥利司他 (14, orlistat) 是胰脂酶的不可逆抑制剂, 阻止脂肪酸甘油酯的水解, 避免了脂肪的同化。
奥利司他分子中的长链模拟脂肪酸的烷基链, 与酶发生疏水-疏水相互作用。四元环的β-内酯具有开环的张力, 但不是很强的酰化基团。酶活化部位的Ser162残基, 在邻近的Asp176和His263的协助下 (形成三元体), 提高了Ser162的亲核性, 加大了该羟基与酰基的电位差, 亲核进攻开环形成稳定的共价结合, 丝氨酸残基被酰化而失活[11], 如图 3所示意。结构要点是β-内酯片段的位置利于三元体对Ser162的助力。虽然是共价键药物, 但未发现脱靶作用。腹泻和脂肪便的不良反应是由于肠内脂肪不能消化吸收的缘故。
 | 图 3 奥利司他与胰脂酶共价结合的示意图 |
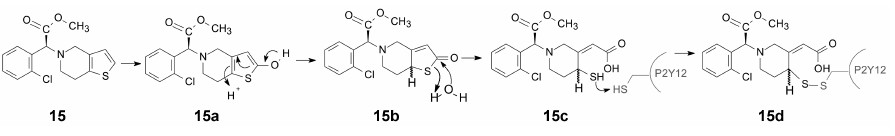
氯吡格雷 (15, clopidogrel) 是血小板聚集抑制剂, 用于预防冠状动脉和脑血管血栓的形成, 其作用机制是被代谢酶CYP2C19氧化代谢, 噻吩2位被羟基化 (15a), 异构化成二氢噻吩酮 (15b), 水解开环生成的巯基丁烯酸, 为活化代谢产物 (15c), 后者与血小板膜上P2Y12的半胱氨酸巯基生成二硫键化合物 (15d), 表现为不可逆的共价键结合[12, 13]。
氯吡格雷是需经代谢活化的前药, 噻吩的2位碳原子要有足够的电荷密度被氧化。这个生化反应过程可借鉴到设计其他的新药分子, 即构建潜在的能与酶分子的半胱氨酸形成二硫化合物, 这在药物合成上是容易实现的, 难点在于设计的含噻吩环的化合物需要能被CYP代谢酶氧化激活, 这具有不确定性, 因为机体对药物或外源性物质的处置遵循最小费力原则, 如果有更容易清除的代谢位点, 肝脏未必按照预先设计的机制进行。
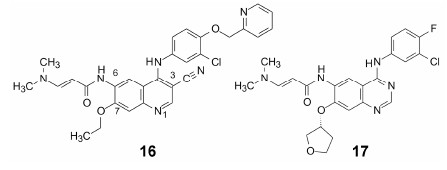
5.4 发生迈克尔加成的抑制剂一些含有α, β不饱和酰胺或酯的药物可与靶标的亲核基团发生麦克尔加成反应, 由于双键与羰基的共轭关系, 加成反应的最终结果是β碳原子对靶标的亲核性原子发生了烷化反应。与芳环相连的丙烯酰胺片段由于电荷的离域化, 是较弱的亲电加成基团, 只能对亲核性强的巯基反应, 难以同羟基或氨基发生迈克尔加成, 这就减少了药物的脱靶作用。当在适宜的空间距离存在巯基 时, 可与巯基形成共价键结合, 这已是生长因子受体酪氨酸激酶抑制剂的惯用片段, 已有数个作为抗肿瘤药物上市。奈拉替尼 (16, neretinib) 和阿法替尼 (17, afatinib) 是表皮生长因子受体 (EGFR) 酪氨酸激酶抑制剂, 作为治疗小细胞肺癌药物已经FDA批准上市。图 4是奈拉替尼与EGFR激酶复合物的晶 体衍射投影图。N1与Met801的NH发生氢键结合, 3位氰基作为氢键接受体与Ser783羟基形成氢键 (阿法替尼则是N3经结构水分子与Ser783形成氢键), 4位的胺苯基结合于疏水腔中。这些结合位点是可逆性EGFR抑制剂 (如厄洛替尼和拉帕替尼等) 的结 合方式。6位的氨基被丙烯酸酰化, 为麦克尔基团, 与EGFR开口处的Cys805距离接近, 发生加成反应, 成为不可逆抑制剂。如果将丙烯酰胺片段移至7位, 则因不适的距离难以发生共价键结合, 活性降低[14]。
 | 图 4 奈拉替尼与EGFR激酶复合物的晶体衍射投影图 |
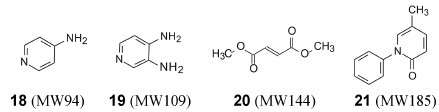
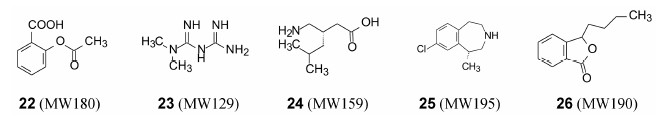
基于靶标结构的分子设计 (SBDD) 常常为满足结合部位的互补要求而将药物分子做大, 虽然提高了对靶标的活性, 却带来药代与物化性质的损失, Lipinsiki的类药5规则(RO5) 对口服小分子药物作了结构的诸多限制, 以对成药性加以保障。如果换一个思路, 能否在超小分子 (分子量低于200) 的化合物空间中寻找创新药物, 也有可能创建一个新的领域。近年来FDA批准的数个超小分子药物已作了诠释。例如阻断钾通道, 增加肌肉神经接头Ach释 放的达方吡啶 (18, dalfampridine) 和阿米吡啶 (19, amfampridine), 用于治疗多发性硬化病和肌无力综合征; 口服治疗牛皮癣和多发性硬化病的富马酸二甲酯 (20, dimethyl fumarate); 抑制转化生长因子β (TGF-β) 的吡非尼酮 (21, pirfenidone) 治疗特质性肺纤维化等。
联想到前已述及的阿司匹林 (22)、2型糖尿病全程一线药物的二甲双胍 (23)、缓解脊髓损伤引起的神经病理性疼痛的普瑞巴林(24, pregabalin)、5-HT2C激动剂减肥药罗卡西林 (25, lorcaserin) 以及抗脑缺血药物丁苯酞 (26, N-butylphthalide) 等等, 对于超小分子药物确实不容忽视。这些药物结构简单, 物化性质好, 对活性和选择性一旦达标, 成药性无多障碍, 而且对生物药剂学的容纳性也很强。
新药创制是人类最复杂的智力活动之一, 周期长, 风险大。每个药物的创制都是个性化的分子操作, 各自都有难以复制的研发轨迹。将活性化合物转化成药物的过程, 几乎是在没有规律性、没有周期性变化的混沌系统中进行的。尽管如此, 由于研发者在研究目标、策略、路径、技术和方法具有趋同性, 突破性和颠覆性的创新较少 (典型的例子是最近相继上市的数个“列净”药物, 都是以抑制SGLT2为靶标、以根皮苷为先导物研发的, 即使是保密和独立进行, 上市的药物结构和药理活性很相近)。在这样的大环境下, 基于靶标和化合物的杂泛性 (promiscuity), 通过反向思维而标新立异, 或许可辟出新的路径。
| [1] | Munos B. A forensic analysis of drug targets from 2000 through 2012[J]. Clin Pharmacol Ther, 2013, 94:407-411. |
| [2] | Breslin HJ, Diamond CJ, Kavash RW, et al. Identification of a dual δ OR antagonist/μ OR agonist as a potential therapeutic for diarrhea-predominant Irritable Bowel Syndrome (IBS-d)[J]. Bioorg Med Chem Lett, 2012, 22:4869-4572. |
| [3] | Guo ZR. Iluxadoline:developed based on peptide ligands[J]. Acta Pharm Sin (药学学报), 2015, 50:1068-1072. |
| [4] | Chen X, Zhao C, Liu L, et al. Terazosin activates Pgk1 and HSP90 to promote stress resistance[J]. Nat Chem Biol, 2015, 11:19-25. |
| [5] | Perry M, Sanguinetti M, Micheson J. Revealing the structural bases of action of hERG potassium channel activator and blocker[J]. J Physiol, 2010, 588:3157-3167. |
| [6] | Kang J, Chen X, Wang H, et al. Discovery of a small molecule activator of the human ether-a-go-go-related gene (HERG) cardiac K+ channel[J]. Mol Pharmacol, 2005, 67:827-836. |
| [7] | Zhou J, Augelli-Szafran CE, Bradley JA, et al. Novel potent human ether-a-go-go-related gene (hERG) potassium channel enhancers and their in vitro antiarrhythmic activity[J]. Mol Pharmacol, 2005, 68:876-884. |
| [8] | Gerlach AC, Stoehr SJ, Castle NA. Pharmacological removal of human ether-a-go-go-related gene potassium channel inactivation by 3-nitro-N-(4-phenoxyphenyl) benzamide (ICA-105574)[J]. Mol Pharmacol, 2010, 77:58-68. |
| [9] | Itoh Z, Omura S. Motilide, a new family of macrolide compounds mimicing motilin[J]. Dig Dis Sci, 1987, 32:915. |
| [10] | Omura S, Tsuzuki K, Sunazuka T, et al. Macrolides with gastrointestinal motor stimulating activity[J]. J Med Chem, 1987, 30:1941-1943. |
| [11] | Hadvary P, Sidler W, Meister W, et al. The lipase inhibitor tetrahydrolipstatin binds covalently to the putative site serine of pancreatic lipase[J]. J Biol Chem, 1991, 266:2021-2027. |
| [12] | Pereillo JM, Maftouh M, Andrieu A, et al. Structure and stereochemistry of the active metabolite of clopidogrel[J]. Drug Metab Dispos, 2002, 30:1288-1295. |
| [13] | Cattaneo M. ADP receptors:inhibitory strategies for antiplatelet therapy[J]. Drug News Perspect, 2006, 19:253-259. |
| [14] | Wissner A, Overbeek E, Reich M, et al. Synthesis and structureactivity relationship of 6,7-disubstituted 4-anilinoquinoline-3-carbonitriles. The design of an orally active, irreversible inhibitor of the tyrosine kinase activity of the epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor-2(HER-2)[J]. J Med Chem, 2003, 46:49-63. |