 2013, Vol. 4
2013, Vol. 4引用本文 |

| 硅钨杂多酸制备、表征及催化油酸酯化反应合成生物柴油活性研究 |

近年来,为了缓解石油短缺和环境保护带来的巨大压力,清洁替代燃料的开发和应用步伐大为加快.在这些替代燃料中,具有环境友好和可再生等优点的生物柴油,已经受到了国内外的广泛关注.大力开发生物柴油这一可再生清洁燃料,是解决能源危机,减少环境污染,走可持续发展道路的重要途径.我国能源植物油资源种类丰富多样,可作为生物柴油生产原料的有麻疯果油、桐籽油、大豆油、蓖麻籽油、棉籽油等.如能加快这些能源植物油作为生物柴油生产原料的综合利用开发步伐,不仅可显著促进区域经济的发展,而且也将对我国新能源战略的实施,发挥重要作用[1-4].
生物柴油的主要成分是长链脂肪酸单酯,其可由上述能源植物油(主要成分为甘油三酸酯)为原料,通过与不同种类的低碳醇(主要为甲醇)进行酯交换反应来制备.当前,欧美各国通常以纯植物油为原料来生产生物柴油[5-6].而在中国,食用油价格过高、食用优先和大量需要进口的现状决定了当前我国很难以精炼植物油为原料,而只能以高酸值废弃油脂(脂肪酸含量可接近90 %)为原料来生产生物柴油,如餐饮废油和皂脚酸化油等[7-8].由于我国产生的废油脂已经达到450万t/a[9],如能将这些废油脂合理利用于生产生物柴油,将具有非常大的经济环保效益.
生物柴油的制备方法可分为物理法和化学法.物理法包括直接混合和微乳液法,化学法包括高温热裂解、超临界、化学酸碱或生物酶催化酯交换法[10-13].通常通过采用化学法来制备得到一种性能优良并且能在使用过程中保持稳定的生物柴油产品.其中,高温裂解法过程简单,没有任何污染物产生,缺点是在高温下进行,需要催化剂,裂解设备昂贵,反应过程所需能耗高,反应程度很难控制,并且当裂解混合物中的硫、水、沉淀物处于规定范围内时,其灰分、碳渣和浊点往往会处于规定值以外.另外,热裂解过程中除去了植物油中所含的氧,而使其不再具有含氧燃料的一些环保方面的优点.
采用超临界法时,经超临界处理的甲醇能在无催化剂存在的条件下与油脂发生酯交换反应,其产率高于普通的催化反应,同时还可避免使用催化剂所必需的分离过程,使酯交换过程更加简单、安全和高效.该方法的缺点是:反应需在高温高压下进行,对反应设备有很高的要求,实现工业化还需进一步研究.酶催化受原料油中游离脂肪酸和水分的影响较小,具有产品分离容易、反应条件温和、醇用量少、甘油易回收且无废物产生等优点.但是,以酶为催化剂时,一旦醇的加入量过多,则会使酶失去活性,影响反应进程,因此对工艺条件的控制较为严格,并且酶在制备时较麻烦,且价格较贵,使其生产成本相对其它几种催化剂来说较高,另外当脂肪酶处于游离状态时,不易回收,因此限制了其在工业上大规模用于生产生物柴油.
化学法当中,均相强碱如KOH催化的酯交换法是目前采用较多的工业化制备生物柴油的方法.虽然该技术比较成熟,但是对原料油的要求高(水分含量 < 0.06 %、游离脂肪酸含量 < 0.5 %)[14].采用酸催化法制备生物柴油时,用于催化油脂与甲醇酯交换反应的液体酸催化剂通常有硫酸、盐酸和苯磺酸.均相强酸如H2SO4催化的酯交换反应可避免皂化反应,对原料的要求不高,但反应结束后需对催化剂进行中和处理,由于含盐废水的产生,造成了一定程度的环境污染;对反应设备有较大的腐蚀性;并且酸催化法制备而成的生物柴油中会有一些酸残留,这些残留酸会对柴油机的金属部件带来一定的腐蚀作用[15].因此,寻找一种环保替代工艺流程显得较为重要.新工艺的重点是合成一种具有高活性、高选择性和高稳定性的固体酸催化剂.
与传统液体酸催化剂相比,近些年来,固体酸催化剂已经成为催化法制备生物柴油的新发展趋势[16-18].固体酸作为生物柴油合成的催化剂,具有以下优势:不易失活,对油脂品质要求不高,能催化转化酸值和含水量较高的油脂,尤其适合以废餐饮油为原料生产生物柴油,且具有易分离、无污染和可循环使用等优势.其中,杂多酸(Heteropoly Acid,简写为HPA)以其强酸性、高活性、对设备无腐蚀、对环境友好、可回收、能重复使用等优点已经受到了研究者的广泛关注.杂多酸是由杂原子(如P、Si、Fe、Co等)和多原子(如Mo、W、V、Nb、Ta等)按一定的结构通过氧原子配位桥联组成的一类含氧多酸,其具有高催化活性和稳定性,且对环境无污染,是一种具有广泛应用前景的绿色催化剂.目前,已报道的可用于催化酯化反应的杂多酸催化剂主要有:H3PW12O40、H4SiW12O40、H3PMo12O40和H4SiMo12O40[19-21].
与传统催化剂相比,杂多酸具有独特的优越性: ①杂多酸的阴离子结构稳定,兼具酸性和氧化性,可以通过改变组成元素(配位原子、中心原子及反荷离子等),来调变其性能,从而满足反应的需要;②通常溶于极性溶剂,反应在均相和多相体系中都能进行.在多相状态下杂多酸为选择性催化剂,在均相条件中杂多酸可用水萃取出来再重复利用或作它用;③独特的反应场.杂多酸具有像沸石一样的笼状结构,其杂多阴离子间有一定空隙,能吸收极性分子进入固体内形成假液相.假液相行为的存在使催化反应不仅在杂多酸表面进行,也可以扩展到其分子内,从而使杂多酸具有更高的活性和选择性.
本研究采用硝酸酸化-乙醚萃取法制备了一种硅钨杂多酸催化剂,并研究了所制备的硅钨杂多酸在催化脂肪酸与甲醇酯化反应时的活性,在进行酯化反应研究时,考虑到油酸是植物油脂中含有的一种主要脂肪酸.因此,在酯化反应研究过程中,以油酸代替脂肪酸进行了催化剂活性研究实验,综合考察了催化剂用量、反应时间、反应温度等对酯化反应产品收率的影响.
1 实验 1.1 试剂实验所用试剂:甲醇(CH3OH, 99.5 %),油酸(CH3(CH2)7CH=CH (CH2)7COOH, 99.5 %),Na2SiO3· 9H2O (CH3OH, 99.5 %),(NH4)2WO4·2H2O (分析纯),乙醚((C2H5)2O, 分析纯),硝酸(HNO3, 分析纯),氢氧化钠(NaOH, 分析纯),无色酚酞溶液,实验用水均为蒸馏水.
1.2 仪器与测试通过XL30型扫描电子显微镜(SEM,荷兰飞利浦)观察了所制备而成的硅钨杂多酸催化剂的整体形貌;通过天津先权公司的tp-5080全自动多用吸附仪测定了催化剂样品的酸位(NH3-TPD),具体步骤如下:在吸附管中装载50 mg测试样品后,打开N2钢瓶,控制流量约为30 mL/min,待流量稳定后升温至300°C,保温30 min后降至室温.关N2钢瓶,打开NH3钢瓶(10 mL/min,0.1 MPa)吸气20 min,关NH3钢瓶,用pH测定尾气,至中性后,打开N2钢瓶,吹扫30 min,调基线归零,控制升温速率为10°C/min,升至800°C;通过Miniflex型X射线衍射仪(日本理学公司)对催化剂样品进行了XRD衍射分析;德国NETZSCH公司的STA409 C/3/F型热重及差热分析仪测定了催化剂样品的热重(TGA)曲线,测试时所使用的气氛为氮气;Nicolet 6700型傅立叶变换红外光谱仪测定了催化剂样品的红外光谱(FTIR)图;PW2424型X荧光分析仪(荷兰帕纳科公司)对催化剂样品进行了荧光分析(XRF);样品比表面积(BET)的测定在美国Quantachrome公司的NOVA4000型自动物理吸附仪上进行.将样品在350°C下脱气2 h后, 以N2为吸附质于77 K恒温吸附.比表面积由N2吸附等温线结合BET方程求得.
1.3 硅钨酸催化剂制备制备硅钨酸的实验步骤如下:称取50 g (NH4)2WO4·2H2O溶于水后加入Na2SiO3·9H2O搅拌使之溶解,加热至沸腾回流.用分液漏斗向上述混合溶液中滴加HNO3至无沉淀产生为止,停止加热;将混合液体用布氏漏斗趁热抽滤,待冷却至室温后移入分液漏斗中,加大量乙醚洗涤;分离后,将产品放入恒温干燥箱烘干,即得产品硅钨酸(H8SiW12O40),其制备原理如反应方程式(1)所示:
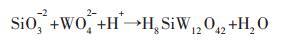
|
(1) |
通过使用低温常压实验装置,进行了硅钨酸催化油酸与甲醇酯化反应研究.反应过程如下:在已连接好冷凝回流管、机械搅拌装置和温度计的250 mL三口烧瓶中按既定比例加入油酸、甲醇和催化剂,加热至所需温度后,开始搅拌计时,反应开始.常压反应的实验条件如下:反应温度(50~70 °C)、催化剂/反应物质量比为(0.2 wt%~1.7 wt%)和反应时间(1~4.5 h).每间隔一定时间,连续取样分析计算油酸的酸值来确定其转化率.采用国标GB/T5530-2005中所介绍的方法来测定反应物的酸值。过程如下:准确称取1 g样品注入锥形瓶中,加入10 mL乙醚-乙醇混合液(体积比为1:1)中,用0.1 mol/L KOH标准溶液滴定,以酚酞为指示剂。另作一空白试验,除不加生物柴油外,其余操作同上,记录空白试验中KOH的用量。酸值计算公式如式(2)所示:
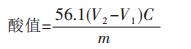
|
(2) |
式(2)中V1为空白试样消耗KOH标准溶液的体积,mL;V2为试样消耗KOH标准溶液的体积,mL;C为氢氧化钾浓度,mol/L;m为试样质量,g.
原料油酸的酸值经测定为144.4 mgKOH/g油脂.通过原料油酸的初始酸值及在不同反应时间所取样品的酸值,可计算得到油酸在不同反应时间下的转化率.通过比较不同反应温度、催化剂/反应物质量比、反应时间下的油酸转化率,可得到硅钨酸催化剂具有最佳活性的反应参数条件.油酸转化率计算公式如式(3)所示:
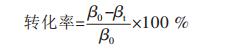
|
(3) |
式(3)中βO为原料油酸的初始酸值;βt为反应时间为t时所取样品的酸值.
2 结果与讨论 2.1 催化剂表征结果图 1为硅钨酸催化剂的扫描电镜(SEM)分析图.由图 1可知,制备而成的硅钨酸催化剂,是一种多孔并且孔径较大的固体催化剂.当一种固体酸催化剂被用于催化制备生物柴油时,由于生物柴油制备过程中涉及大分子脂肪酸的反应,催化剂的孔径是影响其催化活性的一个非常重要的因素.Lopez等的研究工作中,报道了β分子筛的微孔性(
 |
| 图 1 硅钨酸的扫描电镜(SEM)分析图 |
图 2为硅钨酸催化剂的NH3-TPD图.由图 2可知,分别在130 ℃和780 ℃附近出现了2个脱附峰.这两个脱附峰,均属于B酸位,分别对应着一个强酸位和一个弱酸位,弱酸位和强酸位的出现,与孔径和酸位所处的位置有关.弱酸位,主要是由于氨分子在孔中的相互作用;强酸位,主要是由于氨分子与催化剂孔壁的相互作用.由此可知硅钨酸催化剂具有明显的弱酸中心和强酸中心.并且,由于硅钨酸催化剂具有高密度的强酸位.因此,其有望在催化油酸酯化反应制备生物柴油过程中,具有高催化活性.
 |
| 图 2 硅钨酸的NH3-TPD分析图 |
用红外光谱对杂多酸进行表征是最常使用的测定研究方法,一些文献已经指出杂多酸阴离子在FTIR指纹区出现特征峰的主要波数范围为700~1100 cm-1.图 3为制备而成的硅钨酸催化剂的FTIR谱图.由图 3可知,在该区域内出现了4个特征峰,分别为1010、965、904、756 cm-1,分别为Si-O、W-O、W-Ob1-W、W-Ob2-W键的伸缩振动频率.
 |
| 图 3 硅钨酸FTIR谱图 |
图 4为制备而成的硅钨酸催化剂的XRD谱图.由图 4可知,合成的硅钨杂多酸晶型衍射峰出现在2θ=10 °、16 °、26 °、37 °、53 °处,与文献报道结果相一致[23],表明在制备而成的硅钨酸催化剂中,已经形成了硅钨酸晶体.
 |
| 图 4 硅钨酸的XRD谱图 |
图 5为制备而成的硅钨酸催化剂的差热分析图.由图 5可知,硅钨酸在100 ℃左右开始失重,对应着自由水的脱出,失重最快也最多,随着温度进一步升高,热重曲线无明显峰型出现,这也表明制备而成的硅钨酸催化剂具有较好的热稳定性.
 |
| 图 5 硅钨酸的差热分析图 |
通过X荧光分析仪对制备而成的硅钨酸催化剂进行了元素含量分析,其结果如表 1所示.从表 1可知,制备出的硅钨酸中的钨、硅、氧元素的含量分别为48.975 %、2.763 %、20.132 %.由于不同的BET比表面积,将导致催化剂的外表面与内表面具有不同数量的酸位,也将对催化剂的活性产生影响。通过多点N2吸附与脱附法(液氮,-196 ℃),测量了制备而成的硅钨酸催化剂的BET比表面积,为8 m2/g.结合XRD谱图、差热分析、红外光谱和NH3-TPD分析的分析结果,可推断出本研究合成的硅钨酸催化剂有望在用于催化酯化反应时具有高活性和稳定性.
| 表1 硅钨酸的荧光分析结果 |
 |
| 点击放大 |
2.2 催化活性研究结果
当油酸与甲醇的质量比为2:3,反应温度为60 ℃,反应时间为1 h,通过改变催化剂用量对油酸转化率大小的影响进行了研究,结果如图 6所示.从图 6可知,当催化剂用量为反应物的总质量的1.4 %时,转化率最大,为77.84 %.
 |
| 图 6 催化剂用量改变对油酸转化率大小的影响 |
当油酸与甲醇的质量比为2:3,反应时间为1 h,催化剂用量为1.4 %时,研究了反应温度的变化对油酸转化率大小的影响,结果如图 7所示.从图 7可知,反应温度为65 ℃时,催化剂催化活性最高,即油酸转化率最大,为88.17 %.而甲醇的沸点是64.8 ℃,且本次实验得到的最适宜的反应温度为65 ℃.表明在常压条件下进行的甲醇与油酸酯化反应的最佳温度点在甲醇沸点附近.
 |
| 图 7 反应温度的变化对油酸转化率大小的影响 |
当油酸与甲醇的质量比为2:3,反应温度为65 ℃,催化剂用量为1.4 %时,进行了反应时间的变化对油酸转化率大小的影响研究,结果如图 8所示.从图 8可知,当反应时间为3.5 h时,催化活性最高,转化率为96.45 %.
 |
| 图 8 反应时间的变化对油酸转化率大小的影响 |
3 结论
本研究采用回流酸化法和乙醚萃取法制取了一种硅钨酸催化剂,应用多种表征手段对合成的催化剂进行了分析,并研究了其在催化甲醇和油酸酯化反应时的活性.当油酸与甲醇的质量比为2:3,催化剂用量为反应物总质量的1.4 %,反应温度为65 ℃,反应时间为3.5 h的条件下,合成的硅钨酸催化剂具有最佳活性,油酸的转化率最高,为96.45 %.另外,合成的硅钨酸催化剂可回收并循环利用,对环境污染小,是一种理想的可用于高效催化转化高脂肪酸含量油脂为生物柴油的绿色环保型催化剂.
| [1] |
Mustafa B. Potential alternatives to edible oils for biodiesel production-A review of current work[J].
Energy. Convers. Manage, 2011, 52: 1479–1492. DOI: 10.1016/j.enconman.2010.10.011. |
| [2] |
Amish P V, Jaswant L V, Subrahmanyam N. A review on FAME production processes[J].
Fuel, 2010, 89: 1–9. DOI: 10.1016/j.fuel.2009.08.014. |
| [3] | 熊道陵, 舒庆, 李英, 等. 生物柴油催化合成研究进展[J]. 江西理工大学学报, 2012, 33: 10–17. |
| [4] |
Sharma Y C, Singh B, Upadhyay S N. Advancements in development and characterization of biodiesel: A review[J].
Fuel, 2008, 87: 2355–2373. DOI: 10.1016/j.fuel.2008.01.014. |
| [5] |
Georgogianni K G, Kontominas M G, Pomonis P J, et al. Conventional and in situ transesterification of sunflower seed oil for the production of biodiesel[J].
Fuel. Process. Technol, 2008, 89: 503–509. DOI: 10.1016/j.fuproc.2007.10.004. |
| [6] |
Sousa L L, Lucena I L, Fernandes F A N. Transesterification of castor oil: Effect of the acid value and neutralization of the oil with glycerol[J].
Fuel. Process. Technol, 2010, 91: 194–196. DOI: 10.1016/j.fuproc.2009.09.016. |
| [7] |
Bhatti H N, Hanif M A, Qasim M, et al. Biodiesel production from waste tallow[J].
Fuel, 2008, 87: 2961–2966. DOI: 10.1016/j.fuel.2008.04.016. |
| [8] |
Peng B X, Shu Q, Wang J F, et al. Biodiesel production from waste oil feedstocks by solid acid catalysis[J].
Process. Saf. Environ. Protect, 2008, 86: 441–447. DOI: 10.1016/j.psep.2008.05.003. |
| [9] |
Balat M, Balat M, Kirtay E, et al. Main routes for the thermo-conversion of biomass into fuels and chemicals. Part 1: pyrolysis systems[J].
Energy. Convers. Manage, 2009, 50: 3147–3157. DOI: 10.1016/j.enconman.2009.08.014. |
| [10] |
Goering C E, Schwab A W, Campion R M, et al. Soyoil-ethanol microemulsions as diesel fuel[J].
Trans. ASAE, 1983, 26: 1602–1604. DOI: 10.13031/2013.33808. |
| [11] | 贺芹, 徐岩, 滕云, 等. 华根霉全细胞脂肪酶催化合成生物柴油[J]. 催化学报, 2008, 29: 41–46. |
| [12] |
Lee J S, Saka S. Biodiesel production by heterogeneous catalysts and supercritical technologies: review[J].
Bioresour. Technol, 2010, 101: 7191–7200. DOI: 10.1016/j.biortech.2010.04.071. |
| [13] |
Gui M M, Lee K T, Bhatia S. Feasibility of edible oil vs. waste edible oil as biodiesel feedstock[J].
Energy, 2008, 33: 1646–1653. DOI: 10.1016/j.energy.2008.06.002. |
| [14] |
Shi W, He B, Ding J, et al. Preparation and characterization of the organic-inorganic hybrid membrane for biodiesel production[J].
Bioresour. Technol, 2010, 101: 1501–1505. DOI: 10.1016/j.biortech.2009.07.014. |
| [15] |
Feng Y, He B, Cao B, et al. Biodiesel production using cation-exchange resin as heterogenous catalyst[J].
Bioresour. Technol, 2010, 101: 1518–1521. DOI: 10.1016/j.biortech.2009.07.084. |
| [16] |
Zabeti M, Wan M, Wan D. Activity of solid catalysts for biodiesel production: a review[J].
Fuel. Process. Technol, 2009, 90: 770–777. DOI: 10.1016/j.fuproc.2009.03.010. |
| [17] |
Sunita G, Devassy B M, Vinu A, et al. Synthesis of biodiesel over zirconia -supported isopoly and heteropoly tungstate catalysts[J].
Catal. Commun, 2008, 9: 696–702. DOI: 10.1016/j.catcom.2007.08.007. |
| [18] |
Park Y M, Lee J Y, Chung S H, et al. Esterification of used vegetable oils using the heterogeneous WO3/ZrO2 catalyst for production of biodiesel[J].
Bioresour. Technol, 2010, 101(s): 59–61. |
| [19] | 赵忠奎, 李总石, 王桂茹, 等. 杂多酸催化剂及其在精细化学品合成中的应用[J]. 化学进展, 2004(4): 620–630. |
| [20] | 胡长文, 王恩波, 梁虹. 杂多酸的催化技术进展[J]. 现代化工, 1992(4): 36–40. |
| [21] | 莫琪, 蒋治良. 杂多酸催化剂在酯化反应中的新应用[J]. 广西化工, 1998(4): 36–40. |
| [22] |
Lopez D E, Goodwin J G, Bruce D A, et al. Transesterification of triacetin with methanol on solid acid and base catalysts[J].
Appl. Catal. A, 2005, 295: 97–105. DOI: 10.1016/j.apcata.2005.07.055. |
| [23] | 于淑萍, 姚培正, 邓宇. 杂多酸催化剂的制备及应用[J]. 天津化工, 1996(1): 22–24. |