 1992, Vol. 6
1992, Vol. 6引用本文 |

| 几种新型两性捕收剂合成及性能推测 |
两性捕收剂是指那些分子中既含有阴离子基团又含有阳离子基团的药剂。由于其浮选性能优异,特别是在盐类矿物浮选中表现出很好的选择性。因此, 在国内外受到普遍的关注。至今已得到研究的两性捕收剂主要有两种类型。即氨基羧酸类
RNH(CH2)nCOOH
和酰氨基羧酸类
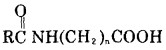
|
这些药剂在磷灰石、孔雀石、氧化铅锌矿等浮选中取得了较大的成功[1~3]。但对于两性捕收剂具有较好选择性和捕收能力的原因, 一直存在较大争议, 人们多从这些药剂的电性行为进行探讨和猜测, 未有满意的结论[4]。
我们提出了另一系列两性捕收剂, 即氨基膦酸类捕收剂。我们曾报道了这类药剂对萤石、方解石、重晶石、白钨矿和磷灰石的浮选性能, 这类药剂表现出比氨基羧酸类两性捕收剂更佳的性能[5~6]。在本文中, 我们将报道这类药剂的结构及合成方法。共有四个化合物, 其名称、分子式和代号如下:
LN11-1 α-苯氨基苄基膦酸
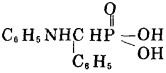
|
LN11-2 α-苄氨基苄基膦酸
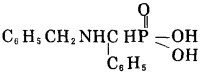
|
LN10-2 β-辛氨基乙基膦酸
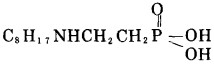
|
LN14-1 亚磷酸单β-二丁氨基乙基酯
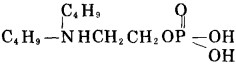
|
为了找到两性捕收剂具有较好浮选性能的本质原因, 我们还进行了药剂结构理论分析和量子化学计算。
2 合成 2.1 α-氨基烃基膦酸α-氨基烃基膦酸的合成有两种方法。其一是以α-卤代烃基膦酸酯或其他带活泼的α离去基的烃基膦酸酯为原料与胺反应获得C-N键的方法
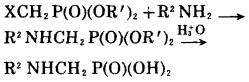
|
(1) |
X=Br.Cl等
该方法由于α-取代膦酸酯xCH2P(O)(OR')2本身制备不容易, 因而不常用。另一合成方法是三配价磷化合物与C=N双键加成, 可以用三配价磷化物与Schiff碱反应或者与醛和胺直接作用, 这是Mannich型反应。此法步骤少, 反应条件温和, 本文合成α-氨基烃基膦酸采用了这一方法, 反应式如下
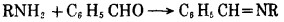
|
(2) |
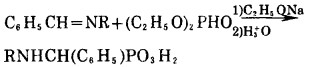
|
(3) |
α-苯氨基苄基膦酸
向装有搅拌器、温度计和回流冷凝器的150ml三颈瓶中, 投入37.1g(0.4mol)新鲜蒸馏的苯胺及42.4g(0.4mol)新鲜蒸馏的苯甲醛, 猛烈搅拌混合, 反应放热,有水生成,反应液静置15分钟, 猛烈搅拌下倾入90ml无水乙醇中, 静置, 有淡黄色结晶析出, 过滤并以乙醇重结晶, 经真空干燥, 得62.0g苄叉苯亚胺固体,熔点52℃, 收率85.5%。然后向装有搅拌器、温度计、回流冷凝器(带CaCl2干燥管)的100ml三颈瓶中, 投入l2.0g(0.08mol)亚磷酸二乙酯, 7.2g(0.04mol)上述制得的苄叉苯亚胺和10ml无水乙醇, 加入2ml饱和乙醇钠的乙醇溶液, 于100℃搅拌反应3小时, 然后室温放置48小时, 有无色结晶生成, 经过滤, 无水乙醇洗涤并干燥,得8.5gα-苯氨基苄基膦酸二乙酯结晶, 将此产物溶解在50ml浓盐酸中, 加入2mlDMF(二甲替甲酰胺), 并在带回流冷凝器的100ml烧瓶中回流24小时, 生成大量兰色固体, 经过滤, 并在乙醇中重结晶, 得6.17g兰色固体α-苯氨基苄基膦酸, 总收率59.0%。以标准Na0H水溶液滴定, 其含量大于95%, pka13.19, pka2 9.74;红外光谱IR(cm-1, 压片, kBr):3400 (N-H伸缩), 2900-2300(P-O-H伸缩), 1156(P=O伸缩), 1600、1490、1451 (苯C=C伸缩), 1300(C-N伸缩)。
α-苄氨基苄基膦酸
向带搅拌器、温度计和回流冷凝器的150ml三颈瓶中, 投入60ml无水乙醇, 15.9g (0.15mol)新鲜蒸馏的苯甲醛, 16.0g(0.15mol)新鲜蒸馏的苄胺, 混合回流10分钟后, 加入100ml蒸馏水, 分出有机相并溶入100ml乙醚中, 以无水氯化钙干燥, 过滤, 减压除去乙醚, 减压蒸馏, 收集142-144℃/666.6Pa馏分, 得24.3g淡黄色液体苄叉苄亚胺, 收率82.6%, 然后, 向装有搅拌器、温度计、回流冷凝器(带CaCl2干燥管)的100ml三颈瓶中, 投入18.1克(0.1mol)苄叉苄亚胺、27.6g(0.2mol)亚磷酸二乙酯和20ml无水乙醇混合液, 加入2ml饱和乙醇钠的乙醇溶液,于100℃回流3小时, 冷却至室温, 用3×20ml蒸溜水洗涤, 有机层用150ml二氯甲烷稀释, 经无水硫酸钠干燥过夜, 减压蒸除溶剂二氯甲烷, 残留物在250ml带回流冷凝器的烧瓶中与100ml浓盐酸回流24小时, 用薄膜蒸发器除去低沸物, 得到的白色固体在乙醇-水中重结晶, 得25gα-苄氨基苄基膦酸产品, 收率90.0%, 含量大于96%, pka14.17, Pka29.86;红外光谱IR(cm-1, 压片、kBr):3400(N-H伸缩), 2870-2250(P-O-H伸缩), 1158(P=O伸缩), 1616, 1500, 1456(苯C=C伸缩), 1310(C-N伸缩)。
2.2 β-氨基烃基膦酸β-氨基乙基膦酸是一类重要的有机化合物, 已有十一个以研究者姓名命名的合成方法, 但这些方法的收率一般不超过50%, 关于β-取代氨基乙基膦酸的报道甚少, Myers于1957年曾报道过碘化三甲铵基乙基膦酸的合成, 路线见式(4)。收率为29%。
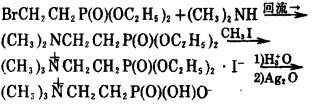
|
(4) |
Kittredge等人报道过另一条合成路线, 以β-氨基乙基膦酸与甲醛缩合并氢化还原, 收率为40%。
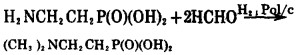
|
(5) |
显然, 此方法也不具备制备价值。本文提出了一个新的简便方法, 以三乙胺作溴化氢的束缚剂, 胺与β-溴代乙基膦酸酯作用得到β-氨基乙基膦酸酯, 再水解成相应的β-氨基乙基膦酸, 此法反应条件温和、操作简单、收率高于以往的方法, 达80%。
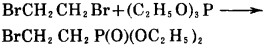
|
(6) |
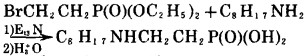
|
(7) |
β-辛氨基乙基膦酸
向装有搅拌器、温度计、短颈冷凝器(带轻组分收集器、氯化钙干燥管和计泡器)的500ml三颈瓶中, 加入48.6g(0.3mol)亚磷酸三乙酯、220.8g(1.2mol)1.2-二溴乙烷, 氮气保护下, 加热至160-165℃,搅拌反应3小时, 收集到流出的轻组分溴代乙烷24.2g。将反应液冷却至室温, 减压抽除轻组分, 减压蒸馏, 收集108-110℃/666.6Pa馏份, 得40.2g无色透明液体β-溴代乙基膦酸二乙酯, 收率55.0%。然后向装有搅拌器、温度计、滴液漏斗和回流冷凝器(带CaCl2干燥管)的150ml四颈瓶中, 放入13.1g(0.1mol)辛胺, 10.1g(0.1mol)三乙胺和50ml干燥石油醚, 搅拌并滴加24.5g(0.1 mol)β-溴代乙基膦酸二乙酯, 有白色固体溴化三乙铵盐析出,反应放热, 控制加料速度使反应温度小于35℃, 加毕,继续搅拌1小时, 放置过夜、过滤, 滤饼以3×15ml石油醚洗涤, 合并滤液, 减压抽除轻组分和溶剂, 然后在150℃/6.66pa下保持20分钟, 得固体β-辛氨基乙基膦酸二乙酯粗产物, 将此粗产物放入100ml烧瓶中(带回流冷凝器), 以50ml浓盐酸与之回流15小时, 然后减压抽去盐酸, 得到的白色固体粗产物经乙醇重结晶, 得β-辛氨基乙基膦酸18.8g, 收率80.0%、含量98.3%, Pka13.03, pka27.73, 红外光谱IR(cm-1、压片、kBr):3400 (N-H伸缩), 2900-2200(P-0-H伸缩), 1280-1180(P=O伸缩)
2.3 亚磷酸单β-氨基乙基酯合成单烷基亚磷酸酯较早的方法是用醇与亚磷酸进行酯化反应
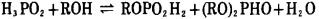
|
(8) |
早期以乙醇、乙二醇为原料进行反应, 由于效果不佳使该方法被认为前途不大。后来通过加入氢氧化钡使反应转化率提高, 该法才又逐渐引起人们的重视。
另一条制备亚磷酸单烷基酯的途径是对二烷基亚磷酸酯进行水解, 水解过程可被酸或碱催化
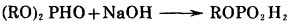
|
(9) |
烷基二氯磷ROPCl2水解也可以制取单烷基亚磷酸酯, 如式(11)。该方法收率较高, 但对反应温度的要求严格, 考虑到原料二丁氨基乙醇的性质, 本文合成亚磷酸单β-氨基乙基酯采取了此法。
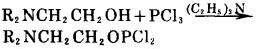
|
(10) |
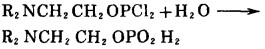
|
(11) |
亚磷酸单二丁氨基乙基酯
向装配有搅拌器、温度计、滴液漏斗和回流冷凝器(带CaCl2干燥管)的250ml四颈瓶中加入13.7g(0.1mol)三氯化磷和150ml沸程为30-65℃的石油醚, 以干冰浴冷却, 于-20℃以下搅拌并滴加17.3g(0.1mol)二丁氨基乙醇、滴毕, 于室温下搅拌2小时, 然后加入50ml水, 以5%碳酸氢钠水溶液调节反应液至碱性, 移入分液漏斗, 除去有机层, 水相以1:1盐酸酸化, 然后浓缩至原体积的五分之一, 用2×75ml二氯甲烷萃取, 合并二氯甲烷萃取液, 以无水硫酸钠干燥过夜。经过滤, 减压除去溶剂, 得到17.5g无色粘稠液体亚磷酸单β-二丁氨基乙基酯, 收率73.0%, 含量100%pka13.75, pka28.66, 红外光谱IR(cm-1, 液膜:kBr片): 3319(N-H伸缩), 2800-2500(P-O-H伸缩), 2400(P-H伸缩), 1382(C-N伸缩), 1212(P=0伸缩), 993(P-O-C伸缩)。
3 药剂结构与性能关系盐类矿物和金属氧化矿极性强、金属原子失去外层s电子或p电子与矿物中其他原子形成典型的离子键, 浮选药剂通常是通过静电力物理吸附或形成离子键化学吸附与之作用, 这就要求药剂的键合原子也要具备相当的极性, 药剂对矿物表面金属离子的作用才容易发生。浮选剂基团电负性值可以准确反映其极性大小, 我们计算了这三种药剂的基团电负性值xg, 结果为:
α-氨基烃基膦酸 xg=4.6
β-氨基烃基膦酸 xg=4.6
亚磷酸单β-氨基乙基酯 xg=4.5
可见, 这些药剂都有较大的基团电负性值, 反映出它们具有较大的极性和与金属形成离子键较强能力。根据基团电负性值划分药剂类型原理[7], 用作氧化矿或盐类矿物捕收剂是合适的, 并已在选矿实验中得到证实[5,8]。
为了深入了解这几种新型两性捕收剂分子结构对其浮选性能可能产生的影响, 我们用量子化学的CNDO/2方法对α-氨基烃基膦酸和β-氨基烃基膦酸进行了计算, 采用的计算模型为:
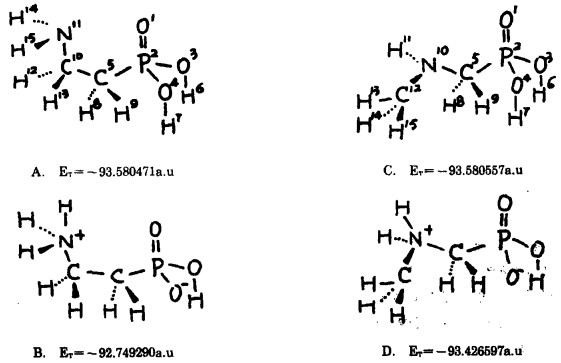
|
其中, ET表示分子总能量。
从这些计算模型各自的总能量可知:α-氨基膦酸以C模型能量最低, 是这类化合物的稳定结构; β-氨基膦酸以模型A最稳定, 能量最低。而它们各自的内盐形式(B、D模型)不是两性捕收剂氨基烃基膦酸的最稳定结构, 这与人们通常的看法不一致。
根据计算结果, α-氨基烃基膦酸和β-氨基烃基膦酸的最高占据轨道可写为:
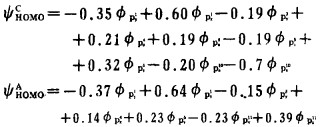
|
可见, 对于β-氨基烃基膦酸A, 分子中氮原子(序号11)对该分子的最高占据轨道有较大贡献, 即氮与经基氧(序号3或4)和膦酰氧(序号l)一道参予了该分子最高占据轨道HOMO的组成。作为配位体, 药剂总是以其最高占据轨道HOMO与金属的最低空轨道<UMO作用成键, 因此, 可以推测, 当β-氨基膦酸与矿物作用时, 分子中N (11)、O(1)、O(3)和O(4)均有可能与矿物金属成键, 这也可以从这些原子所带净电荷看出:
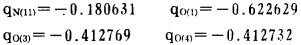
|
但考虑到O(1)与矿物金属作用会有较大的空间位阻, O(3)和O(4)与矿物晶格中金属原子成螯在空间几何方面不是很有利, 而N(11)和O(3)或O(4)与矿物金属原子成六员环螯合物在空间上有利, 因此认为, β氨基烃基膦酸可能是螯合捕收剂, 通过O·N型螯合附着在矿物表面。分析α-氨基膦酸的HOMO组成, 亦存在类似情形, 通过N·O五员环螯合附着在矿物表面, 药剂与矿物的作用模式见图 1。
 |
| (a)α-氨基膦酸 (b)β-氨基膦酸 图 1 氨基膦酸与矿物表面的作用方式 |
由于形成螯合物的空间要求, 这两类药剂应当具备较好的选择性。并且, 从ψHOMOA和ψHOMOC组成还可以看出:β-氨基烃基膦酸的N(11)对其HOMO的贡献比α-氨基烃基膦酸的N(10)对HOMO贡献更大, 这预示β-氨基烃基膦酸更易与矿物形成O·N型螯合物, 可能更具良好选择性。
所有这些推论都已在后来的实验中一一得到了验证[5,6]。
4 结论 4.1α-氨基烃基膦酸通过亚磷酸酯对C=N双键加成可以方便地制得, 亚磷酸单β-氨基乙基酯通过三氯化磷与醇反应再水解可以高收率地被合成; β-氨基烃基膦酸利用本文提出的新方法合成, 产率高、操作简便。
4.2基团电负性计算表明这三种药剂适合用作盐类矿物捕收剂; 通过量子化学计算建立了药剂与矿物作用的N·O螯合模型, 并指出形成螯合物是这几类两性捕收剂选择性优越的本质原因, 且β-氨基烃基膦酸比α-氨基烃基膦酸性能更优。
参考文献略