 1990, Vol. 4
1990, Vol. 4引用本文 |

| 高纯物分析的原子吸收法 |
原子吸收光谱(AAS)是现代分析化学中最有前途的方法之一,它已被用于各种材料的分析。由采用电热原子化的AAS提供的低检测限和足够高的准确度是该法应用于高纯物分析的基础。作为这种目的,AAS的能力胜过发射光谱(ES)和火花源质谱(SSMS)法,见表 1,表中表明了所比较的这三种方法的元素检测限的范围。在许多情况下,AAS给出的检测限与SSMS的一样低,但准确度和精密度优于SSMS, 相对标准偏差,大多数情况AAS是4—20%,ES和SSMS是20—30%。分析15个微量杂质的时间,三种方法几乎相同,但AAS在检查试剂空白,尤其在用标准加入法克服基体效应显得更为方便。
| 表 1 用ES', AAS和SSMS法杂质的绝对检测限 |
 |
| 点击放大 |
我们已经用电热原子化AAS作为一些高纯金属(Ag、As、Au、Ga、In、Re Sn)新的分布方法。
一、 高纯金属的分析AAS的潜在能力并不都能实现,其原因是作用于分析信号上的基体效应,尤其是分析信号的灵敏度大大降低时。今后任何原子吸收手续的开展首先是调查基体影响及消除任何不利因素的方法。在许多情况下,常需预先化学分离除去主元素。
锡和砷的分析。当要分析的基体能易挥发时,就有可能在石墨炉预原子化阶段蒸馏除去干扰基体。我们已在分析高纯锡和砷的新方法中用了此法:锡以四溴化物、砷以氯化物和氧化物挥发掉。这一步要特别注意优化过程,已经确定,干化温度要升到250—300℃, 灰化温度达到900—1000℃,残留在石墨炉中的AS不多于1%,Sn1—5%。低于此灰化温度,这些量将增加,容易干扰痕量分析元素的测定。但另一方面,蒸馏基体可能损失分析元素,所以要折衷考虑。我们考察了不同量的主成份在蒸馏跑掉时痕量杂质的分析信号变化结果,所有杂质的测定都是在预先选择的优化条件,结果表明,当试验的溶液中每毫升达到20mg的As或Sn、Ba、Be、Ca、Co、Cr、Cu、Ni、Sr、V和Yb的测定不受干扰。然而,蒸发基体时,Cd、Fe、Mg或Mn其中任何之一的损失达10-22%以上,测定这些元素就要适当地校正。溶液中As达20mg/ml时,AI、Ca、Co、Cr、Cu、Fe、Li、Mg、Ni、Sr的测定和As达到10mg/ml时,Cd、Pb、Zn的测定都有可能。蒸馏Sn时,微量杂质Al、Bi、Ca、Pb、Sb、Zn以及蒸馏As时Au、Sb、Si和Ga损失达90%, 这些杂质不能用该法测定。
基于上述调查,提出了以下分析高纯锡和砷中杂质的手续,锡样品(100mg)放入PTFE烧杯中,加入1.5ml浓盐酸和0.05mlBr2,在恒温器65—68℃的温度下加热分解锡4—5小时,然后加入去矿质水至5ml,该溶液直接用于分析。砷200mg用1.5ml6M盐酸和0.5ml12M硝酸的混合物加热至70℃恒温分解3—4小时,用去矿质水精确地稀释至10ml,除Pb、Cd、Zn需进一稀释外,所有的杂质都可以直接分析。
测定锡中Ag、Ba、Be、Ca、Cd、Co、Cr、Cu、Fe、Mg、Mn、Ni、Sr、V和Yb,砷中Ag、Al、Ca、Cr、Cd、Co、Cu、Fe、Li、Mg、Mn、Ni、Pb、Sr、Zn。检测限10-7—10-5%,相对标准偏差2—3%。
2.铼的分析。从一种非常难挥发的基体中分馏蒸发出杂质的相对方法被用于高纯铼的分析。用过氧化氢或硝酸分解得到的过铼酸在石墨炉中能被还原为铼(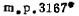
| 表 2 铼中杂质的测定条件* |
 |
| 点击放大 |
杂质浓缩原子吸收分析高纯金属由上注意到,纯物质的直接AAS并不是完全可行,这是由于主成份降低了分析信号的强度。为了消除这种效应,必须把基体与分析元素分离,通常是萃取基体或杂质。
4.铟和镓的分析,由于基体效应;镓中只有Al、Ca、Cu、Mg和Mn能够直接测定,具有足够低的检测限(5×10-8—5×10-5%)。用β.β'一二氯基乙基醚(二氯乙醚)萃取分离铟和镓已经广泛地应用于许多可测定的杂质。水相中基体金属的量要少,不能超过50ug。In和Ga能够用新近蒸馏的二氯乙醚在PH≤1最完全地萃取分离,水相不能含有硝酸盐,因为硝酸镓不能被二氯乙醚萃取。介绍以下分析手续:金属镓(1g)用3ml12M盐酸和1ml14M硝酸混合酸在95°恒温下分解16—18小时。溶液蒸发至呈浆状,加1ml12M盐酸,再蒸一次,使硝酸镓更完全转变为氟络合物,用10ml12M盐酸把残渣转入到分液漏斗中,用3次每次10ml二氯乙醚萃取镓,水相在80°蒸发,用2ml0.3M盐酸分解残渣,该溶液用于分析。金属铟(1g)用4ml0.8M盐酸在60—70°分解。溶液蒸发至干。用5ml8M的盐酸提取残渣并转至烧结硅分离漏斗中。用3次每次10ml二氟乙醚按有机相:水相=2:1的相比萃取分离In。控制溶解时的温度,并利用小体积酸使空白中的杂质含量达到最小。方法已可用于Ga中23个杂质,铟中21个杂质的测定,检测限10-8—10-5%,相对标准差4—30%〔7, 8〕。
5. 金和银的分析。最初研究的在盐酸和硝酸介质中分析上的共同影响表明,在硝酸介质中As、Cd、Co、Cr、Cu、Fe、Ir, Ni, Mn, Mo, Pd, Rh, Sb, Zn, 在盐酸介质甲As、Cd、Co、Cr、Cu、Fe、Ir、Mn、Mo、Ni、Pd、Rh、Zn,每种杂质元素与其它杂质元素的倍比为1 :50(W/W)并不影响测定,但发现在盐酸介质中Ag、Sb、Fe、Bi、Pb、Pt、Se,在硝酸介质中Bi、Pb、Se怨Te有相当大的干扰。为了消除干扰影响,可用硫酸铜(Cu5mg/ml)缓冲,所有被测定的杂质在含有Ag100ug/ml和Au1mg/ml都引起基体影响。为了消除基体影响,用萃取分离基体:银用邻—异丙基—N—甲基胺荒酸盐的氯仿溶液萃取,金用Z—正—丁基硫醚的仿溶液萃取,除Pd外,所有微量杂质都保留在水相中,因此,在分离后的样品中测定Pd:对金,用乙醚萃取分离金;对银,用0.3M二辛基硫醚的甲苯溶液萃取钯。
样品制备如下:金(1—10g)以硝酸和硫酸(1:3)混合酸并加过氧化氢分解,然后反复加入浓盐酸蒸发赶去硝酸,用1M2—正一丁基硫醚的氯仿溶液从2M盐酸中萃取金,蒸发水相得到湿状盐,用0.1M盐酸溶解至体积5—25ml(准确测量),加入硫酸铜后用于Ag、As、Bi、Cd、Pb、Pt、Se和Te的分析。银(0.5)用1.8ml6M硝酸分解,用蒸馏水稀释溶液至5ml, 然后用等体积的2M的邻—异丙基—N—甲基荒酸盐的氯仿溶液萃取,水相用0.2M萃取剂的氯仿溶液洗涤后蒸发,残余物用0.1M硝酸分解,所得溶液加硫酸铜作缓冲剂用于As、Bi、Cd、Pb、Sb、Se、Te的分析,利用缓冲剂改善所有这些元素的灵敏度和精度,否则会引起相互干扰。
带入的Rh和Ir完全进入溶液,利用交流电电解溶解分离样品,Ag和Au中杂质的检测限是1×10-9—2×10-6%。
银和金能用直接法分析F和Si, 方法是基于P和Si与钼形成磷钼酸(PMA)和硅硫酸(SMA),而原子吸收测定其中的钼。金先用乙醚萃取分离,然后用钼酸铵形成PMA, 再用正丁醇和四氯化碳(1:3)的混合物萃取,用3M的氨水提取钼,用AAS测定。由于桂在分解样品时可能转变为非活性的二氧化硅而损失,用AgCl为载体共沉淀硅,随后用氢氧化钠加热AgCl转变为活性形式,过滤,过滤的主溶液用于分析。先用正丁醇和四氯化碳(1:3)的混合物从硝酸中萃取PMA, 然后用异戊醚从硫酸介质中萃取SMA。测定硅,是测量有机相中钼的吸收。测定磷,是在水相中提出的PMA测量钼的吸收。银中P和Si, 金中P的检测限是5×10-7%, 相对标准差≤6%〔11〕。
我们也试验了用石墨炉直接分析固体样品Ag和Au中钯的可能性〔11〕,它可免去样品预处理的时间消耗并在信号上减少干扰元素影响,把10mg样品放在石墨炉,中部的Lvov平台的凹部,标准是用纯度99.9999%的Ag和Au制成的人工合金。对银的分析,用选择的加热条件在分析的原子化之前有可能蒸馏掉80—90%之多的基体。二阶段使用的温度程序是“阶段Ⅰ—在2100°蒸馏基体1—1.5分钟; 阶段Ⅱ—在2650°杂质原子化15—20秒。银中Pt、Pd和Rh的检测限是(2—5)×10-9g,相对标准差<10%,金中Pd和Rh的检测限分别为6×10-9和2×10-8g, 在测定金中Pd和Rh的优化条件下(阶段Ⅰ—1100—1200°30秒,阶段Ⅱ—2650°15—20秒)仅有30—40%的金被蒸掉,因此用一个挖制样品校正残余的基体影响,无干扰元素影响检测。
二、 讨论电热AAS的应用已允许我们发展灵敏的方法用于一些高纯金属在广泛的元素范围分析。在所试验的所有金属中的杂质检测限已概括在表 3中。AAS也使它有可能测定那些在发射光谱中灵敏度差的杂质。这些元素包括P、Si和Hg。“冷蒸发”技术已能测定纯Cd、Ge、In和Te中的汞,检测限~10-6%〔13〕。
| 表 3 无火焰原子吸收测定纯金属中痕量杂质的检测限(%) |
 |
| 点击放大 |
实际,通过更优化的实验条件和用更纯的试剂,有些杂质的检测限还有可能降得更低。
然而,当使用浓缩技术时,又出现一些问题。通常,1g样品用萃取浓缩,电热AAS用于15—20个微量杂质需大约1ml溶液,这样在被分析溶液中的杂质浓度(W/W)与原来材料中的杂质浓度大至相同。用更大的样品重量(达到10g)在相同的溶液体积时可以达到更高的浓度,如上所述的金。然而,这样常常导致试剂空白中杂质量增加,由此检测限也增加。因此,我们宁可用那种加大样量、以最少的试剂消耗的富集方法,这些包括蒸溜、沉淀等等。这些方法的优点可用高纯锡的分析来说明,用四溴化锡蒸馏基体结合电热AAg测定杂质,Ag、Ca、Co、Cr、Fe、In、Mg、Mn、Ni、Pb、Zn的测定检测限可达到1×10-9—5×10-8%之低。
参考文献(略)
许春才译自Talanta, Vol.34, NO.1, PP147—161, 1987
叶鹏鸿、曾怀北校。