 1990, Vol. 4
1990, Vol. 4引用本文 |

| 钪的分离与测定 |
钪属于伴生稀散元素,原子量44.9559, 熔点1200℃,是第三副族元素,具有稳定的三价。钪在化学反应中的行为与铝和稀土有相似之处,钪的氢氧化物与铝的氢氧化物均具有两性,钪的氟化物和稀土氟化物一样是难溶的,但又不同于稀土和钍,氟化钪易溶于过量的碱性金属氟化物中,生成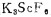
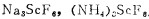
钪只有一种独立矿物—钪钇石(ScY)2SiO7氧化钪35—42%, 但由于钪钇石中钪与钇的分离难度很大,成本昂贵,至今钪钇石未能成为提取钪的主要来源,目前世界上钪主要从铀钍矿、钨锡矿、稀土矿、钛铁矿、铝土矿及白云母等副产品中回收。黑钨矿中氧化钪的含量可达0.1—1.0%(工业指标是0.02%), 特别是大晶体的黑钨矿。我国钨矿产量非常丰富,居世界首位,近年来在钪资源回收方面取得了突破性的进展,这对综合利用钨矿非常有利。
一、 钪的分离钪的分离方法大致可分为四类:
(一)溶剂萃取〔2—6.26〕 是使用最广泛的方法,根据分离元素的不同,采用不同试剂和有机溶剂。在PH5.5的溶液中,用铜试剂——氯仿萃取分离锆、铪、铀(Ⅳ)等易水解的元素,此时钪不被萃取,过量铜试剂存在,可与近95%的钍分离,大部分干扰元素分离后,用0.1M PMBP—苯(或甲苯在PH1.5—6.0(一般采取PH3.0—5.5)溶液中萃取钪和稀土,用PH2.4的甲酸溶液反萃取稀土,然后用5%盐酸反萃取钪。中性磷类萃取剂磷酸三丁酯(TBP)是较好的萃取剂〔7〕,在8—9N盐酸介质中萃取钪,用3—4N盐酸反萃取,可分离铁、锰等干扰元素,在溶盐酸介质中萃取,可与铍、铝、铬(Ⅲ)分离。过氧化氢存在可掩蔽钛,萃取络合物的组成为SnCl3•XTBP, 已为放射性同位素和红外光谱所证实,如在硝酸介质中萃取,萃取络合物的组成为Sc(NO3)3•3TBP。KCNS—乙醚可从0.5N盐酸中萃取钪和钍、稀土、锰等元素分离,此时钴、钼、铼、同时被萃取。铍、铝、铟、铁(Ⅲ)部分被萃取。噻酚甲酰三氟丙酮(TTA)——苯可在PH1.5的溶液中萃取钪与镧系元素及钙等元素分离。PH≥4.5时,苯甲酰苯胲的异戎醇溶液可萃取钪,当含锆和钛时,可在2—6N盐酸介质中用3%的上述萃取剂萃取锆和钛,钪留在水相,铜铁试剂的氯仿溶液可萃取铁、钛、锫等元素,钪留在水相。
(二)沉淀分离〔8〕 用沉淀分离方法分离钪的试剂有酒石酸、碱金属氟化物(NaF•KF), 镧盐和镁盐作共沉淀剂,草酸沉淀可将钪与稀土一起沉淀,而与钛、铁等分离,这两种沉淀方法至今在许多钪的分离过程中经常使用,但往往与其它分离手段结合使用,特别对于低含量的钪,目前尚未发现单独使用沉淀分离使钪与复杂的干扰元素分离的报导。
(三) 离子交换〔9〕 此法主要应用在分离钍和稀土方面。使用的交换树脂有IR-100H, Dowex—50等阳离子交换树脂及Dowex—1,AB—17等阴离子交换树脂。国内常用的有717苯乙烯型强碱性阴离子交换树脂,用0.1M硫酸铵—0.025M硫酸作淋洗液,依Fe++→Mn++→稀土(Y组)→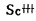
(四) 色层分离〔4、10〕 纸上色层一般用于与稀土元素的分离。展开剂丁酮一乙酸甲酯一硝酸一水的比例85:5:5:5,温度11—32℃, 色层时间1.5—5小时,色层纸必须经15%硝酸铵溶液处理,否则色带不清晰,且干扰元素分离不好。纸上色层多采用国产新华1号快速或中速色层纸。Rf值TR < Sc < Th < u,分别为0.11, 0.63,0.93;萃取色层分离钪是分离效果比较好的一种方法。在4.7—8.7M高氯酸介质中,用TBP作萃取剂,用增水硅胶作载体,经过裝有吸附剂的色层柱后,钪被吸附在柱上,余均不被吸附,因此,大量锆、稀土,铁、铝、钛、铬、铜等元素均可被很好地分离除去,钪可用稀盐酸或水进行洗脱。
二、 钪的测定到目前为止,国外报导测钪的方法中最多的是中子活化(约占70%)。由于仪器、原子反应堆及场地等的限制,国内尚未广泛使用。发射光谱应用较为普遍,亦较为成熟〔11〕,大量铁、钛等元素干扰测定,随着三相萃取应用的日益广泛,微量钪经萃取富集后进行光谱测定是行之有效的方法〔12〕。质谱、电化学,原子吸收等方法报导得不多,成熟的更少〔13〕;光度法是国内测钪的主要方法之一〔4、5、8、9、14、15〕,但遗憾的是没有一个对钪有很好选择性的试剂,都必须预先分离干扰元素,所以方法往往繁杂。化学发光法中,荧光法测钪的报导较多〔16.17.27〕但在所提到的荧光试剂中,大部分是一时无法买到或合成的。因此应用受到了限制。在光度法中,这一情况也常有出现。下面简单介绍一下光度法测钪的几种试剂:
铬天青S在表面活性剂的存在下,与钪生成络合物,过量表面活性剂可分解镧和钇的络合物,因此镧和钇不干扰钪的测定,这一反应在PH5.5及弱碱性溶液中均可进行,前者λmax=625nm,后者λmax=550nm〔18、19〕
漂兰6B(Eriochrome Azurol B简称ECAB)于PH5.80—6.80介质中,在非离子表面活性剂乳化剂OP存在下,与Sc3+形成Sc—ECAB—OP三元络合物,最大吸收波长为659nm,表观摩尔吸光系数为2.43×105,0—11微克Sc/25毫升范围内服从比耳定律,本法测定PH范围较宽,反射度高,灵敏高,线性范围也较宽,活性剂用量少,是一个较好的测钪体系〔29〕。
甲基二甲苯酚兰(methylxlenol blue)在PH2.3—2.7的醋酸钠—盐酸缓冲液中与钪生成络合物,λman= 585nm,共优点是对于铁、锰等干扰元素的分离条件不苛刻,25倍量的锰(Ⅱ)不干扰铁(Ⅱ)可用抗坏血酸还原成铁(Ⅱ),且允许25倍量铁(Ⅱ)的存在,作了40种元素的干扰试验,其中26种不干扰,相对误差 < 5%〔20〕。
1.5—22—(2—羟基—3羧基—5—磺苯基)—3氯基甲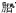
1.5—22—(2—羟基—3、5、6—三氯代苯基)—3—乙酰基甲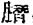
经过一系列分离后,用3—羟基—4—(2—羟基—5—硝基偶氮)—荼—2.7—二磺酸〔Snlphonitaazo R〕在PH2.0的氯化钾—盐酸缓冲液中,在二苯胍存在下,萃取钪于乙醇中,在540纳米处,钪有最大的吸光度。1500倍量的铝,400倍量的镧,100—200倍量的钙、铌、镓,10—50倍量的钍、钒、钛、钨不干扰测定〔21、22〕。
二甲酚橙在PH>1.3的一氯乙酸缓冲液中与钪生成络合物,4—90微克Sc/25毫升服从比耳定律,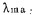
吡啶偶氮间苯二酚(PAR)——铌间接比色法〔8〕,在PH4—6的醋酸——醋酸钠介质中,Sc3+与柠檬酸和铌生成无色三元络合物,未被Sc3+络合的铌与PAR和酒石酸生成鲜红色络合物,钪的含量越高,未被络合的铌越少,溶液的颜色就越浅,从铌—PAR—酒石酸的曲线中可求得钪的含量,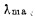
Janamo等用二安替比林甲烷和3—(4—磺苯基偶氮)变色酸(SPADNS)在PH7.6—8.0形成络合物,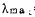
钪和甲基百里酚兰及二苯胍在PH6.5—8.5时形成的络合物用丁醇萃取后可测0.4—40微克/毫升钪,络合物的组成为Sc:甲基百里酚兰:二苯胍=1:2:2〔24〕。
Epeuum等认为8—羟基喹啉是比较好的测钪试剂,PH1.5时,0.2—30微克钪服从比耳定律。
5.7—二硝基—8—羟基喹啉与罗一明B以离子缔合物形式萃取—荧光光度法测钪,检出下限为0.02微克/毫升,方法有较高的灵敏度和低的检出限〔25〕。
由于钪对国民经济的日趋重要,研制出一个准确快速检测钪的分析方法非常必要,近几年来,有机试剂的理论研究正在走向量子化学加电子计算机和激光技术的道路〔28〕,不仅可以解决以前所不能解决的理论问题,而且还可以予言新试剂的性质,进行定向合成,分离和测钪难的问题,必定会得到很好解决。
| [1] |
郭承基.
, 稀有元素矿物学[M]. , 1965.
|
| [2] | |
| [3] |
ЛемРоь Б、Ц、. [J].
《Ж.А.Х》), 1976, 31(12): 2298.
|
| [4] |
雷剑泉, 中科院贵阳地化所, 矿物中微量钪的分离和测定.
|
| [5] |
沈坚. [J].
《江西有色金属》, 1989, 3(1): 49.
|
| [6] |
Desai D.D.. [J].
《Analyst(London)》, 1981, 106(1264): 806. |
| [7] |
шцанкоба Г、Ц. [J].
《Ж.А.Х》, 1978, 33(4): 699.
|
| [8] |
福建省地质中心实验室,吡啶—(2—偶氮—4—问苯二酚铌比色测定钪
|
| [9] |
陈君文, 广东省地质实验所, 矿石中微量钪的分离与测定.
|
| [10] | |
| [11] |
陈城运. ICP发射光谱法测定黑钨矿中钪[J].
有色金属研究院分析年报, 1982: 153.
|
| [12] |
Жцгоппчеп В、П、. [J].
《аоночцаб》, 1972, 33(2): 145.
|
| [13] |
李锡安等, 湖南省地质局实验室, 矿石中微量钪的测定.
|
| [14] |
郑用熙. [J].
《分析化学》, 1983(3): 210.
|
| [15] |
杜士奎, 中科院化工冶金研究所, 钪—铬天生s—溴代十六烷基三甲铵(CTM-AM)三元络合物的形式分光光度法测定钪.
|
| [16] |
Копоненко П、Ц、. [J].
《Ж.А.Х》, 1975, 30(9): 1716.
|
| [17] |
Тдачцпоьцп Ъ、Ь、. [J].
《Ж.А.Х》, 1980, 35(7): 1283.
|
| [18] |
崛内芳藏. [J].
《分析化学》, 1968, 17: 1486.
|
| [19] |
Танама П、Ц、. [J].
《Ж.А.Х》, 1980, 35(2): 279.
|
| [20] |
上田穰一. [J].
《日本化学会志》, 1973, 8: 1467.
|
| [21] | |
| [22] | |
| [23] |
Тамамо П、П、. [J].
《Ж.А.Х》, 1972, 27(2): 261.
|
| [24] |
Е |
| [25] |
Кчаякоьа Ы、Ю、. [J].
《Ж.А.Х》, 1983, 38(6): 1023.
|
| [26] |
Madhri B D. [J].
《Talana》, 1979, 26(9): 892.
|
| [27] |
村田旭. [J].
《分析化学》, 1978, 12: 788.
|
| [28] |
Ечеценко С、Ы、. [J].
《Ж.А.Х》, 1975, 30: 2299.
|
| [29] |
郑用熙. [J].
《化学通报》, 1982(12): 716.
|