 1989, Vol. 3
1989, Vol. 3引用本文 |

| 用三羟基萤光酮类物质光度法测定岩矿中之钨 |
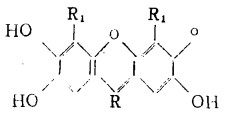
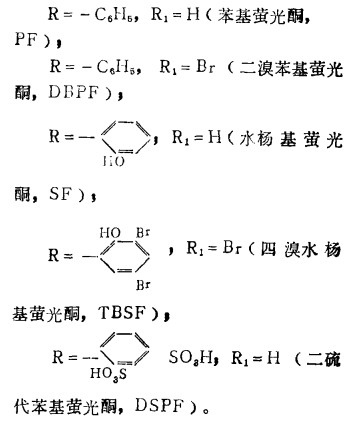
己推荐了用于测定岩浆岩和沉积岩中的钨的各种技术(1—3),采用硫氰酸盐和甲苯—3,4—二硫酚作光度试剂。常常需要用离子交换法、萃取法、或用α—苯偶姻肟、二硫酚沉淀予先富集钨,因为有些方法灵敏度与选择性差,需取较多的试样进行分析。为了改善岩石中钨的光度测定的灵敏度和选择性,研究采用比硫氰酸盐或二硫酚更灵敏的试剂似乎是有益的,我们发现,2,3,7 —三羟基萤光酮和3、4、5—三羟基萤光酮衍生物在表面活性剂存在下与钨有很灵敏的反应,伴有趋向较高酸度的宽广的反应条件。(4—7)但2,3,7—三羟基萤光酮类物质是更灵敏的试剂,在典型阳离子表面活性剂十六烷基吡啶(CP)和十六烷基三甲基铵(CTMA)离子和典型非离子表面活性剂合成丹宁醇(syntanol),三硝基甲苯(Triton)×—100和OC-20存在下,我们已把其研究推广至若干更多的衍生物。所试验的三羟基萤光酮类是:
|
|
采用1×10-3M钨酸钠溶液(在0.5M氢氧化钠中)。0.06M的盐液酸—乙醇溶液中制备PF、SF和DSPF溶液(1-2×10-3M),在1:1(V/V)的水和乙醇混合物中制备DBPF和TBSF溶液(5×l0-4M);这些溶液在使用的当天配制。制备1%的明胶溶液,以及0.01M阳离子表面活性剂溶液,1%和10%非离子表面活性剂溶液和0.01M柠檬酸钠溶液。用盐酸、硫酸、氨水和醋酸铵缓冲溶液调节PH。
(二) 过程把所需数量的酸或缓冲溶液,乙醇、柠檬酸钠,表面活性剂或明胶(按顺序)放入25毫升的容量瓶中,随即放入三羟基萤光酮溶液和钨溶液,加入柠檬酸钠防止钨水解。用等摩尔系列,摩尔比和等吸光点法分光光度测定钨和三羟基萤光酮的结合比。用平衡移动法研究了钨—三羟基萤光酮二元络合物与表面活性剂之间的相互作用。
在白金皿中用浓硫酸(2.0—2.5ml)和浓盐酸(l0ml)分解试样(0.25—0.50克)。试样分解前,含有碳酸盐应用盐酸处理,随即蒸发至干,油页岩和淤泥应在450—500°下灼烧。蒸发酸混合物直至SO3白烟出现,冷却,加入水(2—3ml),并再次蒸发至呈湿盐状。放在水中冷却,用过氧化氢氧化,然后过滤,必要时要用烧结玻璃板漏斗过滤(孔隙度4),用氨水中和至PH5—7,加入浓盐酸(5ml),蒸发至l0ml,再加入1 ml2.5%的在浓盐酸中的Ticl3溶液,并煮沸5—10分钟,往冷却的溶液中加1ml0.4%二硫酚—锌盐络合物乙醇悬浮液,调节盐酸浓度至6M并在水浴上加热10分钟。冷却,加5ml氯仿振荡2分钟。分离提取物,并在白金皿上蒸发至干,然后,在马弗炉中于500°下,灼烧10—15分钟。冷却,加入2ml1%碳酸钠溶液和3滴饱和氯酸钾溶液,蒸发至干,并在750—800°下熔融2—3分钟。用水5-7ml浸出冷却的熔块,加入2.5ml20%盐酸联氨溶液,5ml 5%的甘露糖醇(或山梨糖醇)溶液和2.5ml10%的EDTA溶液。煮沸3—4分钟以使体积减少至10—12ml,并冷却,加入0.5ml0.01M的柠檬酸钠,4ml6M的盐酸,0.5ml5%的抗坏血酸溶液1ml1%氟化铵溶液,1.5ml0.002M的水杨基萤光酮和2ml0.01M的CP或CTMA,在25ml容量瓶中用水稀释至标线,20分钟后,在5—cm比色皿中,在518纳米处,测定相对于经过全部手续的试剂空白的吸光度,由已知量的钨(0—10μg)和试样处理所用的全部试剂(包括:盐酸联氨、甘露糖醇,EDTA,柠檬酸钠、盐酸、抗坏血酸,氟化铵,水杨基萤光酮和CP或CTMA)的吸光度绘制标准曲线。
二、 结 果在明胶(作稳定剂)存在下,钨(V1)与三羟基萤光酮类作用的研究显示出与前人的研究相同,即在酸性介质中,形成了单色的络合物;PF与DBPF作试剂(R),W:R之比是1:2,而SF和TBSF是1 :1。在咕吨环形物中溴化的试剂(即带-Br基的萤光酮衍生物)在较大酸度下起作用(PF和SF为PH2—2.5,而DBPF和TBSF为PH0.5—1.0)。为了使明胶体系稳定,应使乙醇含量达30%。较高的乙醇含量降低吸光度。表面活性剂体系需要较低的乙醇含量。
加入阳离子表面活性剂代替明胶作稳定剂提高络合物生成的酸度范围,对于硫酸盐溶液和DBPF与TBSF, 这个影响是明显的。采用非离子表面活性剂,酸度范围变化较小,但阳离子与非离子表面活性剂对分光光度特性的影响是类似的。在某些情况下,使用非离子表面活性剂产生异常的钨—三羟基萤光酮的络合比。
图 1示出在CP存在下,在不同酸度时,钨—PF络合物的吸收光谱。向该体系加入表面活性剂引起光谱的增色移动,在505—515纳米处出现谱带和在540纳米处的凸出部分,其强度取决于溶液PH和乙醇及表面活性剂的浓度。对所研究的所有三羟基萤光酮类和表面活性剂,观测到类似的结果。
 |
| 2×10-6MW;1×10-5MPF;1×10-3MCP;1×10-4M柠檬酸钠;10%乙醇。2cm比色皿。曲线1和2,试剂空白;1,1MH2SO4;2,PH1.3.曲线:3-7.络合物;〔H2SO4〕,M:3 1.0;4, 0.75 5;0.5;6,0.4;7,0.1. 图 1 在CP存在下,在各种酸度时,苯基萤光酮钨的吸收光谱: |
除了在505—515nm的谱带外,吸收光谱一般不会随表面活性剂浓度而改变,其强度随表面活性剂浓度而增大,直至临介胶团浓度(CMC),然后变成恒定,与表面活性剂浓度无关。图 2示出W—DSPF—CP体系,溶液的表面张力与吸光度对表面活性剂浓度的依赖关系。CP浓度大于CMC时,出现最大吸光度和最小表面张力。
 |
| 2×10-6MW;1×10-5MDSPF;1×10-4M柠檬酸钠;1%乙醇:PH1.0(HCl) 图 2 W—DSPF—CP体系的吸光度(1)和表面张力(2)对溶液中CP浓度的依赖关系: |
为使钨—三羟基萤光酮稳定,必须添加乙醇。用2—10%V/V乙醇获得最大吸光度。在PH3时,该体系只产生一种有色络合物,但硫酸盐介质中的W—SP—CTMA和W—SP—CP体系除外,它产生几种络合物的混合物。在所研究的大多数体系中,尚未证实在广泛的表面活性剂浓度范围内,生成理想配比的三元络合物。所研究的体系的络合物生成条件和络合物的分光光度特性概述于表 1。
| 表 1 在表面活性剂存在下钨与三羧基光酮类络合物的特性 |
 |
| 点击放大 |
讨 论
由实验结果与文献资料比较,可以推断,钨钼与2,3,7—三羟基萤光酮类生成络合物的机理是相似的(8—9)。与钼一样,在表面活性剂浓度为临介胶团浓度(CMC)或大于此浓度时,呈现出三羟基萤光酮钨光吸收特性的最佳结果。这证明表面活性剂胶团在络合物稳定中起决定性作用。胶团可能通过不参与金属离子配位的酸根与三羟基萤光酮分子作用。钨离子与固定在胶团上的萤光酮分子作用。这就是为什么在任何稳定剂(明胶、阳离子表面活性剂,非离子表面活性剂)存在下生成的络合物的光谱是相似的,由于三羟基萤光酮的这种固定作用,防止了钨络合物的凝聚,并且促进了溶液内细分散的吸光颗粒的产生,在吸收光谱中观测到增色移动。对于不同的三羟基萤光酮类和不同的表面活性剂钨—三羟基萤光酮比由1:1至1:4的异常变化,是由于我们还不能完满解释的各种各样相互作用引起的。所研究的三羟基萤光酮类和表面活性剂胶团的大小和疏水性是不同的,而且,表面活性剂胶团的形式、结构和“容量”也不同,它决定着钨(V1)与不同数目的三羟基萤光酮离子配位的可能性。可能是TritonX—100胶团不是刚好与单个PF和SF分子相互作用,而是与它们的氢键聚集体相互作用,这可能是观测到1:4络合物的原因。这也许是证明了这样的观测结果,即组成1:4的络合物不能显示最强的颜色,亦即在表面活性剂存在下,络合物的摩尔吸光度的增大不是由于增大金属离子配位能力引起的。可用阳离子表面活性剂(CP,CTMA)和非离子表面活性剂(Triton X—100,OC—20)使钨三羟基萤光酮稳定。这些表面活性剂存在下形成的钨水杨基萤光酮这种目前熟知的金属与有机分析试剂形成的最灵敏的络合物之一。
可惜,其灵敏性稍差。Nb,Ta,Ti,Zr,Hf,Mo,Sb,Sn和Fe全部干扰。用α—苯偶姻肟或二硫酚预先萃取钨可消除这类元素的若干影响,用EDTA,盐酸联氨和甘露糖醇(或山梨糖醇)混合物可掩蔽它们中的几种元素,尤其是钼。最后加入抗坏血酸和氟化铵以掩蔽由掩蔽剂混合物带入的任一微量的铁(Ⅱ)。测定0.2—1μg,钨的最大允许量是Nb(zoμg),Ta (30),Mo(250),Sn(200)和(mg) Zr(0.5)V(0.5),Cr(> 1),Fe(>4),Cu、Ni、Zn、Mn(10—20)。
本法已用于测定镍基合金和碱金属卤化物与高铼酸盐中的微量钨(10),我们已把本法用于测定打算作为苏联科学院西伯利亚分院地球化学研究所指出的标准试样的若干岩石和泥砂中的钨(表 2)。由分析钠长石化的花岗岩,CT—1A﹝检定值2.3±0.4×10-4%W;测得的钨含量为2.28±0.06×10-4%(95%置信介限),n=21,相对标准偏差6%﹞和镍合金(检定W含量0.035%,测定钨含量为0.0335±0.0004%)的标准试样,确定了本法的有效性。
| 表 2 地质试样中的钨的8次平行测定结果 |
 |
| 点击放大 |
许孙曲译自《Talanta》,Vol.34, No.1 PP.215—218(1987)
余南廉 王丁梁校